Lethbridge Research and Development Centre, 5403 1 Ave South, Lethbridge, AB, T1J 4P4, Canada.
Department of Clinical Sciences, Colorado State University, Fort Collins, CO, 80523, USA.
Sci Rep. 2018 Apr 12;8(1):5890. doi: 10.1038/s41598-018-24280-8.
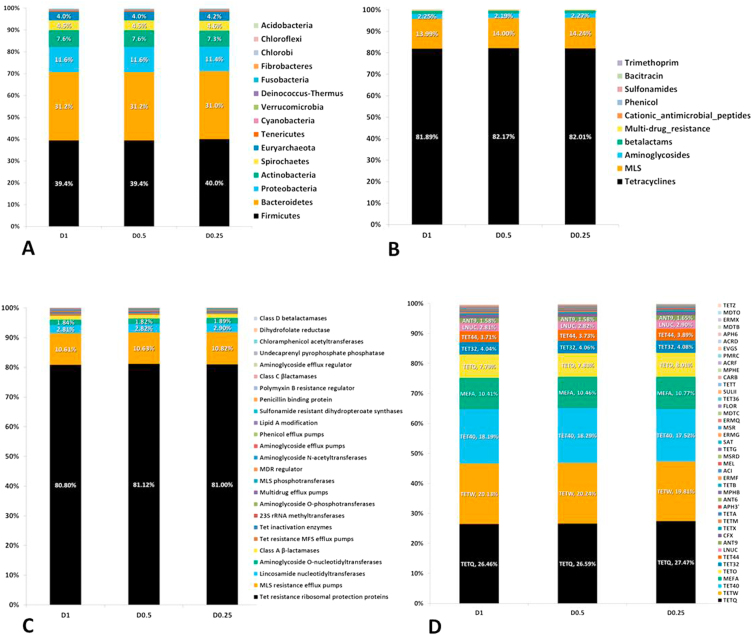
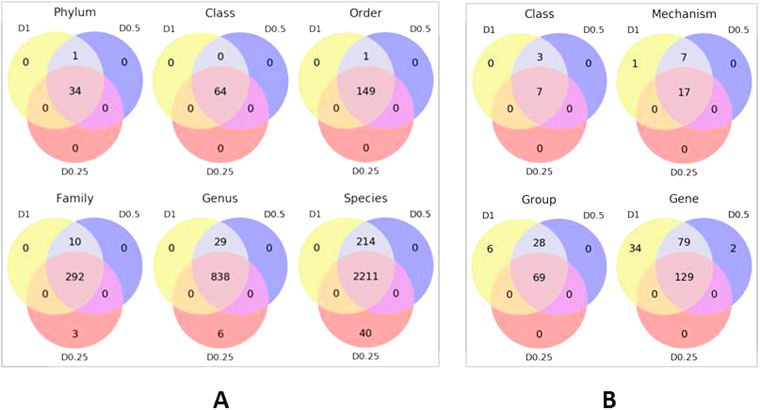
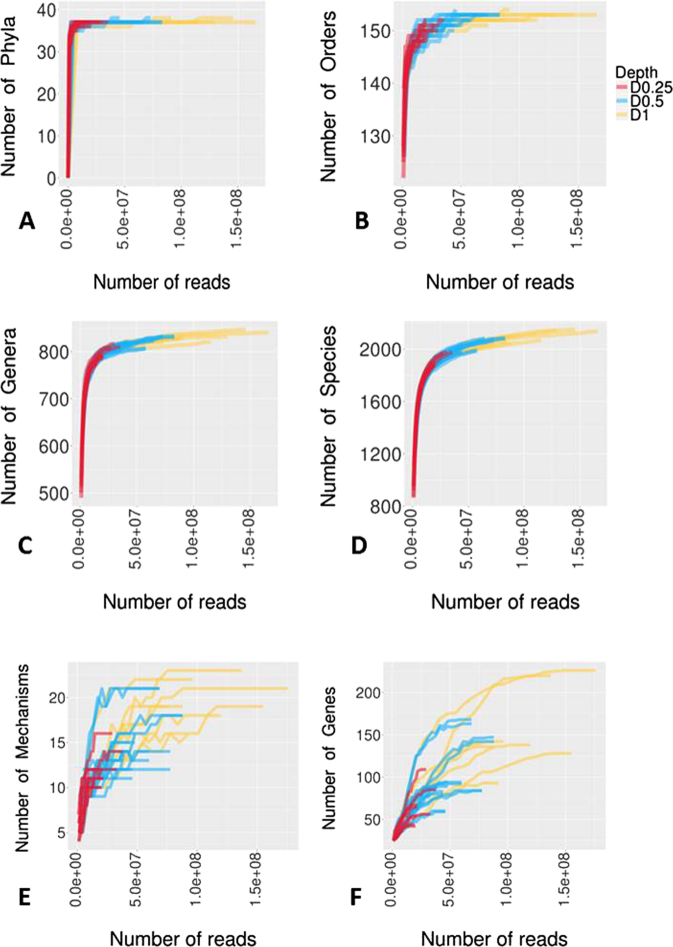
Developments in high-throughput next generation sequencing (NGS) technology have rapidly advanced the understanding of overall microbial ecology as well as occurrence and diversity of specific genes within diverse environments. In the present study, we compared the ability of varying sequencing depths to generate meaningful information about the taxonomic structure and prevalence of antimicrobial resistance genes (ARGs) in the bovine fecal microbial community. Metagenomic sequencing was conducted on eight composite fecal samples originating from four beef cattle feedlots. Metagenomic DNA was sequenced to various depths, D1, D0.5 and D0.25, with average sample read counts of 117, 59 and 26 million, respectively. A comparative analysis of the relative abundance of reads aligning to different phyla and antimicrobial classes indicated that the relative proportions of read assignments remained fairly constant regardless of depth. However, the number of reads being assigned to ARGs as well as to microbial taxa increased significantly with increasing depth. We found a depth of D0.5 was suitable to describe the microbiome and resistome of cattle fecal samples. This study helps define a balance between cost and required sequencing depth to acquire meaningful results.
高通量下一代测序(NGS)技术的发展迅速提高了人们对整体微生物生态学以及不同环境中特定基因的发生和多样性的认识。在本研究中,我们比较了不同测序深度在生成有关牛粪便微生物群落分类结构和抗生素耐药基因(ARGs)流行情况的有意义信息方面的能力。对来自四个肉牛饲养场的八个复合粪便样本进行了宏基因组测序。对宏基因组 DNA 进行了不同深度的测序,D1、D0.5 和 D0.25,平均样本读数分别为 1.17、5.9 和 2600 万。对不同门和抗菌类别的读长比对相对丰度的比较分析表明,读长的相对比例保持相当恒定,与深度无关。然而,被分配给 ARGs 以及微生物类群的读长数量随着深度的增加而显著增加。我们发现 D0.5 的深度适合描述牛粪便样本的微生物组和抗生组。本研究有助于在成本和获得有意义结果所需的测序深度之间找到平衡。