Department of Genetics, Rutgers University, Piscataway, New Jersey, United States of America.
Human Genetics Institute of New Jersey, Rutgers University, Piscataway, New Jersey, United States of America.
PLoS Genet. 2018 Apr 23;14(4):e1007341. doi: 10.1371/journal.pgen.1007341. eCollection 2018 Apr.
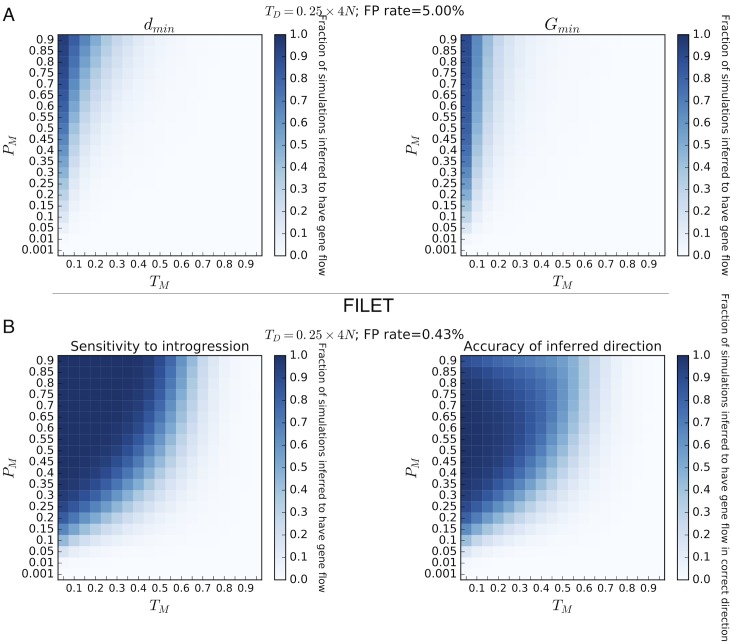
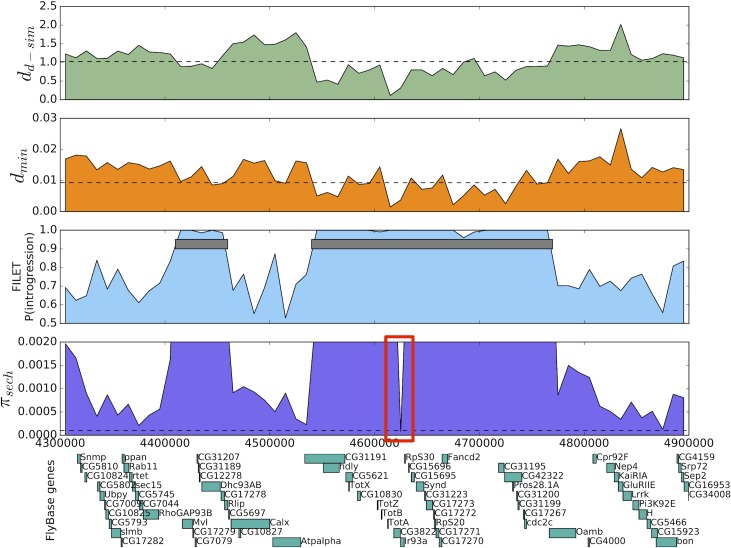
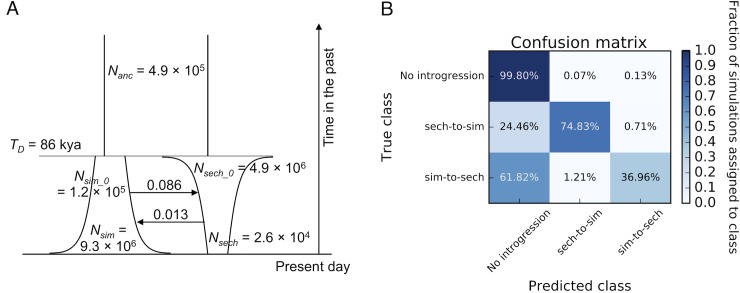
Hybridization and gene flow between species appears to be common. Even though it is clear that hybridization is widespread across all surveyed taxonomic groups, the magnitude and consequences of introgression are still largely unknown. Thus it is crucial to develop the statistical machinery required to uncover which genomic regions have recently acquired haplotypes via introgression from a sister population. We developed a novel machine learning framework, called FILET (Finding Introgressed Loci via Extra-Trees) capable of revealing genomic introgression with far greater power than competing methods. FILET works by combining information from a number of population genetic summary statistics, including several new statistics that we introduce, that capture patterns of variation across two populations. We show that FILET is able to identify loci that have experienced gene flow between related species with high accuracy, and in most situations can correctly infer which population was the donor and which was the recipient. Here we describe a data set of outbred diploid Drosophila sechellia genomes, and combine them with data from D. simulans to examine recent introgression between these species using FILET. Although we find that these populations may have split more recently than previously appreciated, FILET confirms that there has indeed been appreciable recent introgression (some of which might have been adaptive) between these species, and reveals that this gene flow is primarily in the direction of D. simulans to D. sechellia.
种间杂交和基因流动似乎很常见。尽管很明显,杂交在所有被调查的分类群中都广泛存在,但基因渗入的程度和后果在很大程度上仍然未知。因此,开发必要的统计工具来揭示哪些基因组区域最近通过与姊妹种群的基因渗入获得了单倍型是至关重要的。我们开发了一种新的机器学习框架,称为 FILET(通过 Extra-Trees 发现基因渗入的基因座),它能够比竞争方法更有效地揭示基因组的基因渗入。FILET 通过结合来自多个群体遗传综合统计数据的信息来工作,包括我们引入的几个新的统计数据,这些数据捕捉了两个种群之间的变异模式。我们表明,FILET 能够以高精度识别经历了相关物种之间基因流动的基因座,并且在大多数情况下可以正确推断出哪个种群是供体,哪个是受体。在这里,我们描述了一个杂交二倍体果蝇 sechellia 基因组的数据集,并将其与 D. simulans 的数据相结合,使用 FILET 来研究这些物种之间最近的基因渗入。尽管我们发现这些种群的分裂时间可能比以前认为的更近,但 FILET 证实了这些物种之间确实存在大量的近期基因渗入(其中一些可能是适应性的),并揭示了这种基因流动主要是从 D. simulans 到 D. sechellia。