Section of Human Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA.
Department of Pediatric Genetics, Amrita Institute of Medical Sciences and Research Center, Cochin, Kerala, India.
Hum Genet. 2018 Apr;137(4):293-303. doi: 10.1007/s00439-018-1882-3. Epub 2018 Apr 24.
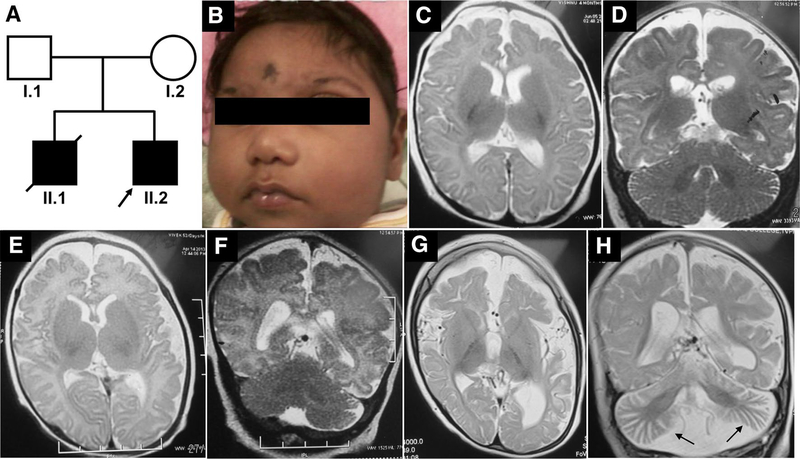
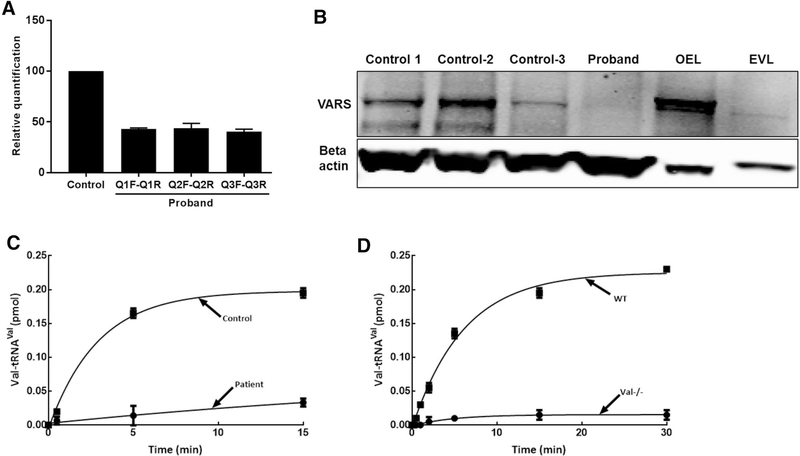
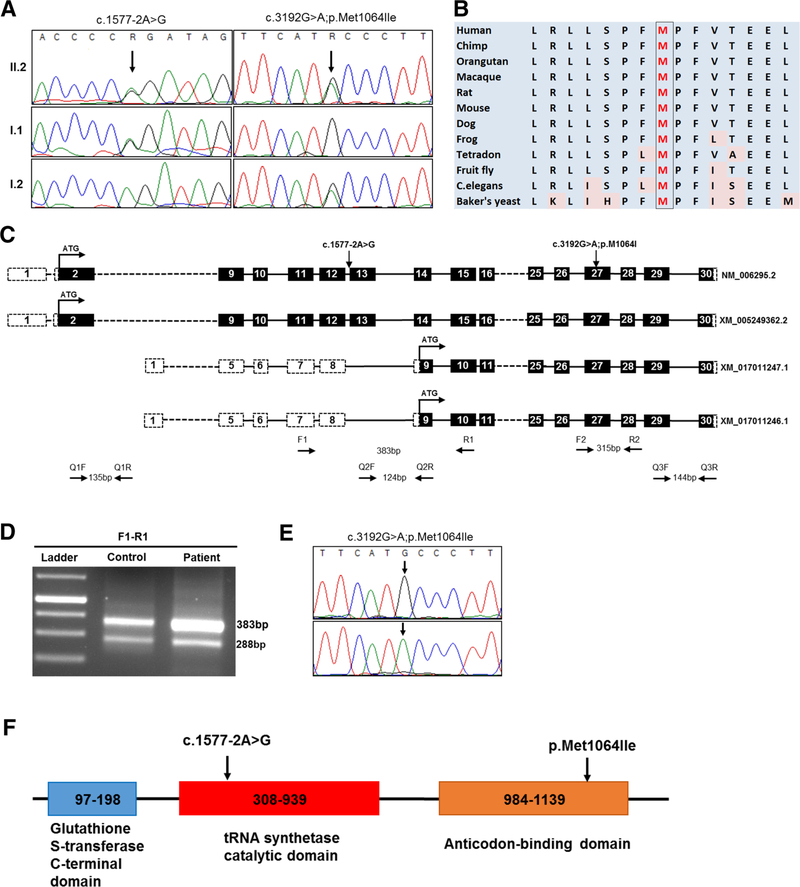
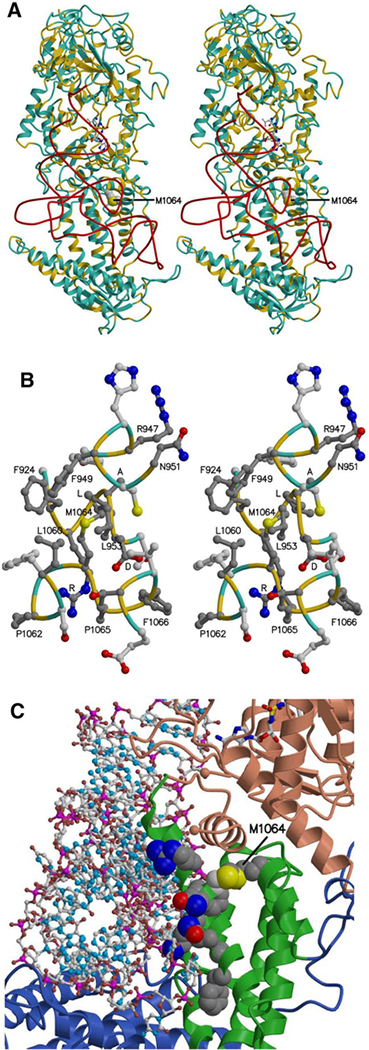
Progressive microcephaly and neurodegeneration are genetically heterogenous conditions, largely associated with genes that are essential for the survival of neurons. In this study, we interrogate the genetic etiology of two siblings from a non-consanguineous family with severe early onset of neurological manifestations. Whole exome sequencing identified novel compound heterozygous mutations in VARS that segregated with the proband: a missense (c.3192G>A; p.Met1064Ile) and a splice site mutation (c.1577-2A>G). The VARS gene encodes cytoplasmic valyl-tRNA synthetase (ValRS), an enzyme that is essential during eukaryotic translation. cDNA analysis on patient derived fibroblasts revealed that the splice site acceptor variant allele led to nonsense mediated decay, thus resulting in a null allele. Three-dimensional modeling of ValRS predicts that the missense mutation lies in a highly conserved region and could alter side chain packing, thus affecting tRNA binding or destabilizing the interface between the catalytic and tRNA binding domains. Further quantitation of the expression of VARS showed remarkably reduced levels of mRNA and protein in skin derived fibroblasts. Aminoacylation experiments on patient derived cells showed markedly reduced enzyme activity of ValRS suggesting the mutations to be loss of function. Bi-allelic mutations in cytoplasmic amino acyl tRNA synthetases are well-known for their role in neurodegenerative disorders, yet human disorders associated with VARS mutations have not yet been clinically well characterized. Our study describes the phenotype associated with recessive VARS mutations and further functional delineation of the pathogenicity of novel variants identified, which widens the clinical and genetic spectrum of patients with progressive microcephaly.
进行性小头症和神经退行性变是遗传异质性疾病,主要与神经元存活所必需的基因相关。在这项研究中,我们研究了一对非近亲家族中两个患有严重早发性神经表现的同胞的遗传病因。全外显子组测序鉴定出 VARS 中的新型复合杂合突变,与先证者共分离:错义突变(c.3192G>A;p.Met1064Ile)和剪接位点突变(c.1577-2A>G)。VARS 基因编码细胞质缬氨酰-tRNA 合成酶(ValRS),是真核翻译过程中必需的酶。对患者来源的成纤维细胞进行 cDNA 分析显示,剪接位点受体变异等位基因导致无义介导的衰变,从而导致无效等位基因。ValRS 的三维建模预测,错义突变位于高度保守区域,可能改变侧链包装,从而影响 tRNA 结合或破坏催化和 tRNA 结合结构域之间的界面。对皮肤衍生成纤维细胞中 VARS 的进一步定量显示,mRNA 和蛋白表达水平显著降低。对患者来源细胞的氨酰化实验显示 ValRS 酶活性明显降低,提示这些突变是功能丧失。细胞质氨酰-tRNA 合成酶的双等位基因突变已知在神经退行性疾病中起作用,但与 VARS 突变相关的人类疾病尚未得到临床充分表征。我们的研究描述了与隐性 VARS 突变相关的表型,并进一步阐明了新型变异体的致病性,这拓宽了进行性小头症患者的临床和遗传谱。