School of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan 646000, P.R. China.
Mol Med Rep. 2018 Jun;17(6):8091-8100. doi: 10.3892/mmr.2018.8895. Epub 2018 Apr 19.
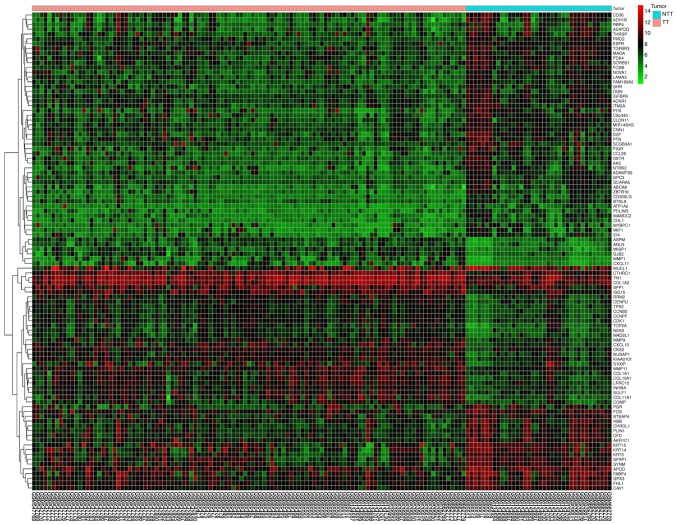
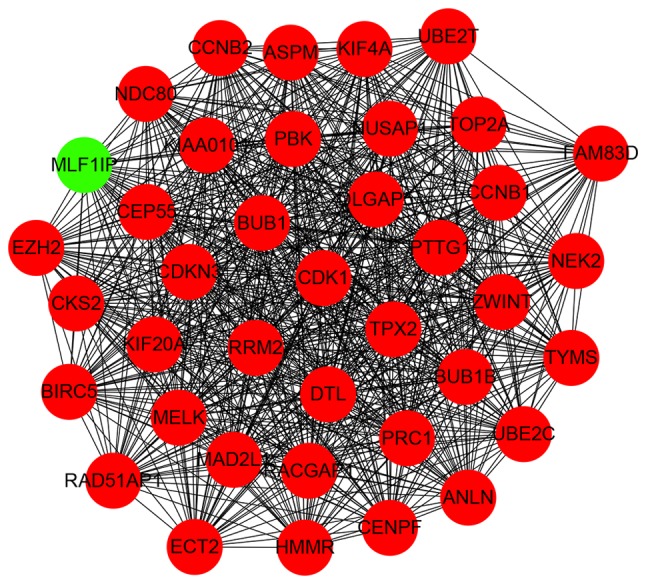
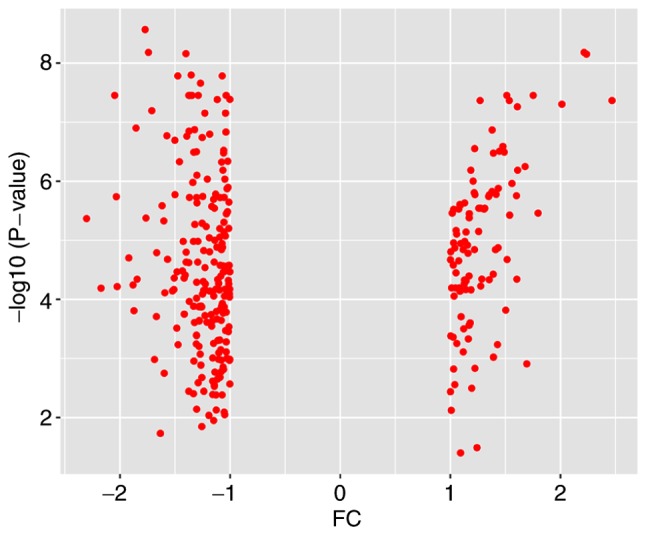
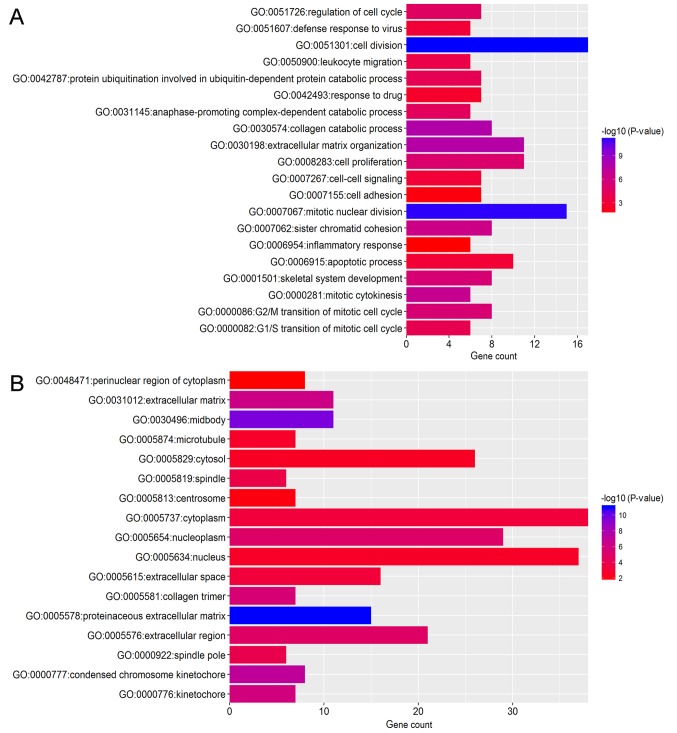
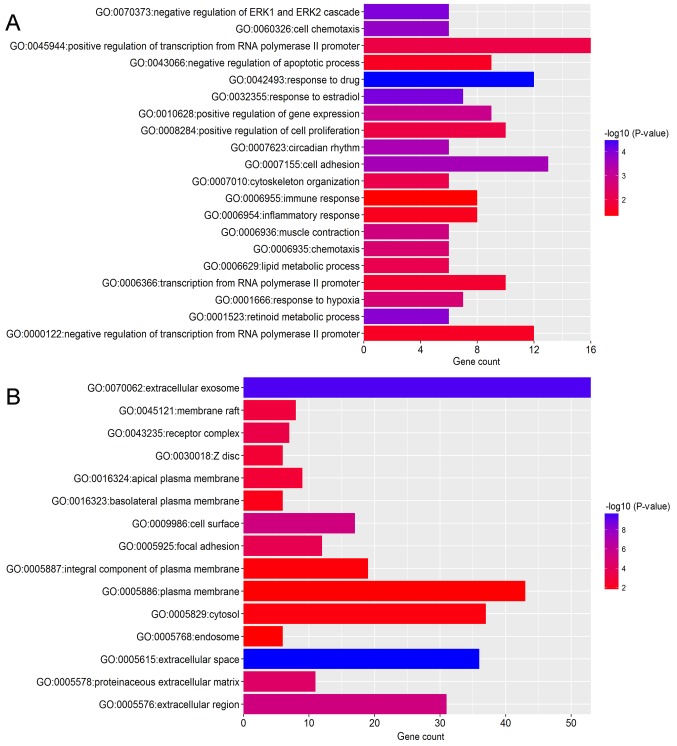
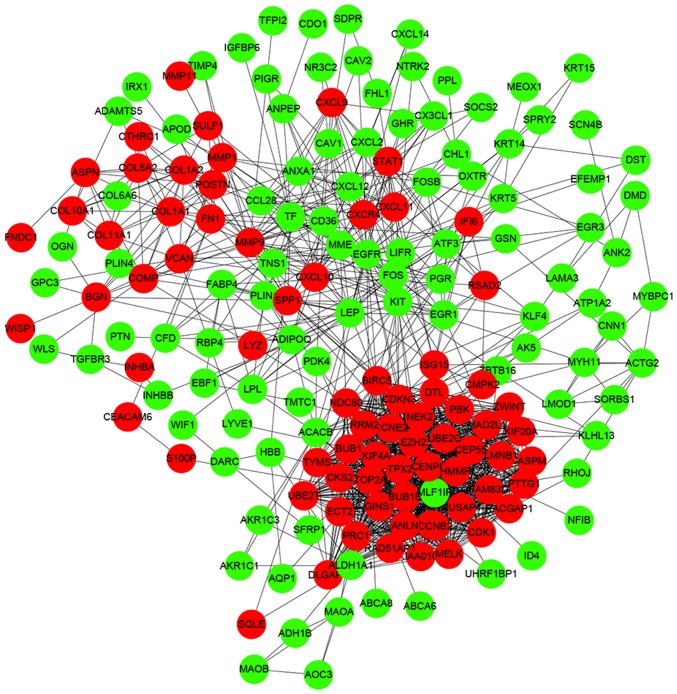
Breast cancer (BC) is the leading malignancy in women worldwide, yet relatively little is known about the genes and signaling pathways involved in BC tumorigenesis and progression. The present study aimed to elucidate potential key candidate genes and pathways in BC. Five gene expression profile data sets (GSE22035, GSE3744, GSE5764, GSE21422 and GSE26910) were downloaded from the Gene Expression Omnibus (GEO) database, which included data from 113 tumorous and 38 adjacent non‑tumorous tissue samples. Differentially expressed genes (DEGs) were identified using t‑tests in the limma R package. These DEGs were subsequently investigated by pathway enrichment analysis and a protein‑protein interaction (PPI) network was constructed. The most significant module from the PPI network was selected for pathway enrichment analysis. In total, 227 DEGs were identified, of which 82 were upregulated and 145 were downregulated. Pathway enrichment analysis results revealed that the upregulated DEGs were mainly enriched in 'cell division', the 'proteinaceous extracellular matrix (ECM)', 'ECM structural constituents' and 'ECM‑receptor interaction', whereas downregulated genes were mainly enriched in 'response to drugs', 'extracellular space', 'transcriptional activator activity' and the 'peroxisome proliferator‑activated receptor signaling pathway'. The PPI network contained 174 nodes and 1,257 edges. DNA topoisomerase 2‑a, baculoviral inhibitor of apoptosis repeat‑containing protein 5, cyclin‑dependent kinase 1, G2/mitotic‑specific cyclin‑B1 and kinetochore protein NDC80 homolog were identified as the top 5 hub genes. Furthermore, the genes in the most significant module were predominantly involved in 'mitotic nuclear division', 'mid‑body', 'protein binding' and 'cell cycle'. In conclusion, the DEGs, relative pathways and hub genes identified in the present study may aid in understanding of the molecular mechanisms underlying BC progression and provide potential molecular targets and biomarkers for BC.
乳腺癌(BC)是全球女性中最常见的恶性肿瘤,但对于参与 BC 肿瘤发生和进展的基因和信号通路知之甚少。本研究旨在阐明 BC 中的潜在关键候选基因和通路。从基因表达综合数据库(GEO)下载了五个基因表达谱数据集(GSE22035、GSE3744、GSE5764、GSE21422 和 GSE26910),其中包含 113 个肿瘤和 38 个相邻非肿瘤组织样本的数据。使用 limma R 包中的 t 检验识别差异表达基因(DEGs)。随后对这些 DEGs 进行了通路富集分析,并构建了蛋白质-蛋白质相互作用(PPI)网络。从 PPI 网络中选择了最显著的模块进行通路富集分析。共鉴定出 227 个 DEGs,其中 82 个上调,145 个下调。通路富集分析结果表明,上调的 DEGs 主要富集在“细胞分裂”、“蛋白质细胞外基质(ECM)”、“ECM 结构成分”和“ECM-受体相互作用”,而下调基因主要富集在“药物反应”、“细胞外空间”、“转录激活剂活性”和“过氧化物酶体增殖物激活受体信号通路”。PPI 网络包含 174 个节点和 1257 个边缘。鉴定出 DNA 拓扑异构酶 2-a、杆状病毒凋亡抑制重复蛋白 5、周期蛋白依赖性激酶 1、G2/有丝分裂特异性周期蛋白-B1 和着丝粒蛋白 NDC80 同源物为前 5 个枢纽基因。此外,最重要模块中的基因主要参与“有丝分裂核分裂”、“中体”、“蛋白质结合”和“细胞周期”。综上所述,本研究中鉴定的 DEGs、相对通路和枢纽基因可能有助于理解 BC 进展的分子机制,并为 BC 提供潜在的分子靶点和生物标志物。