European Molecular Biology Laboratory (EMBL) Hamburg Outstation, DESY, Hamburg, Germany.
Sci Rep. 2018 May 8;8(1):7204. doi: 10.1038/s41598-018-25355-2.
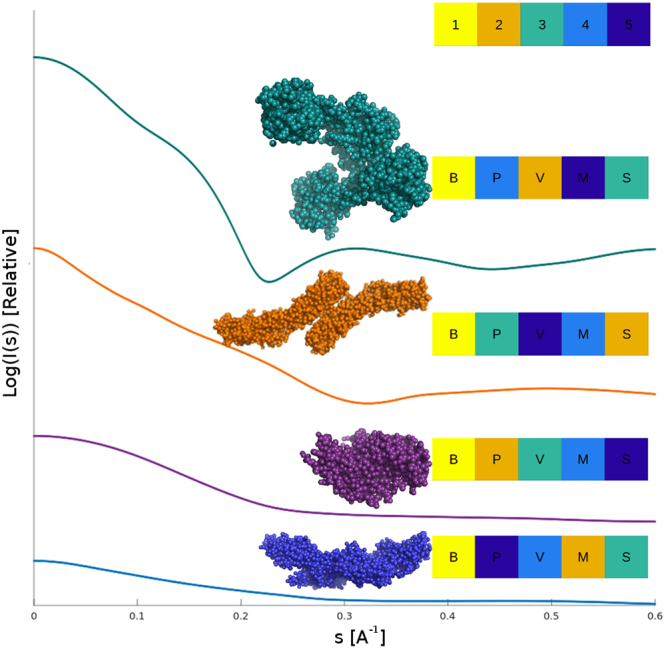
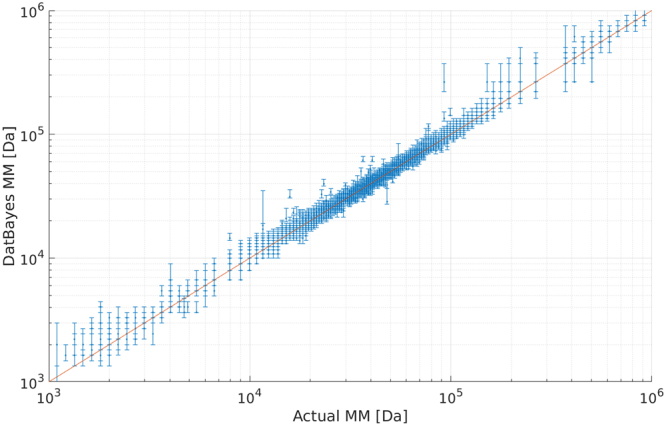
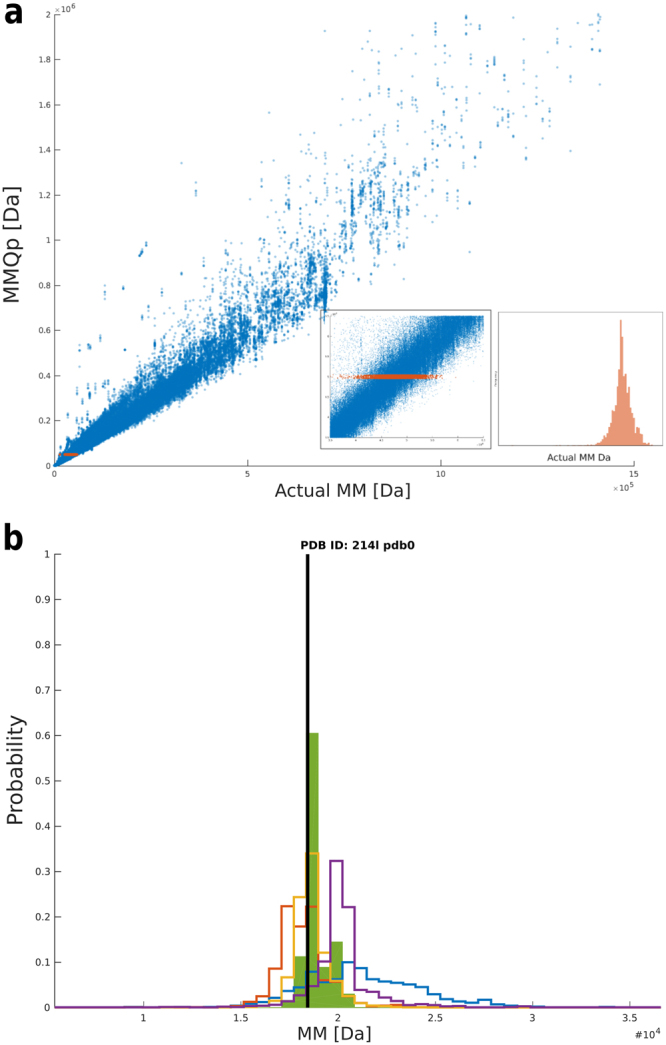
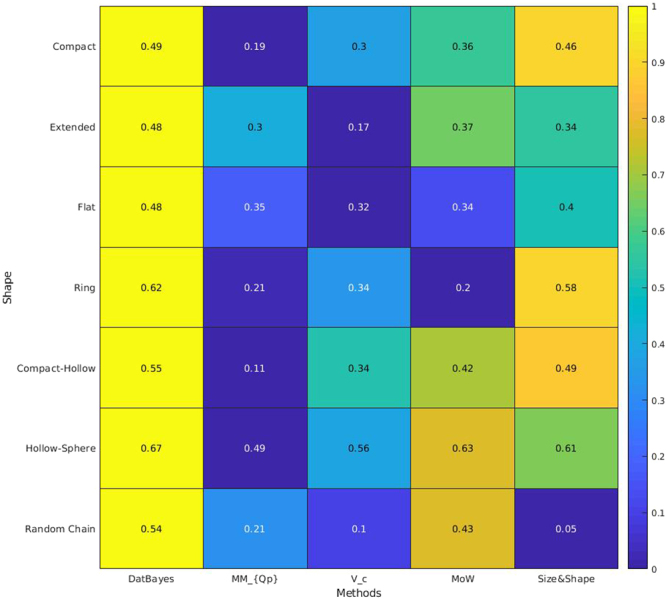
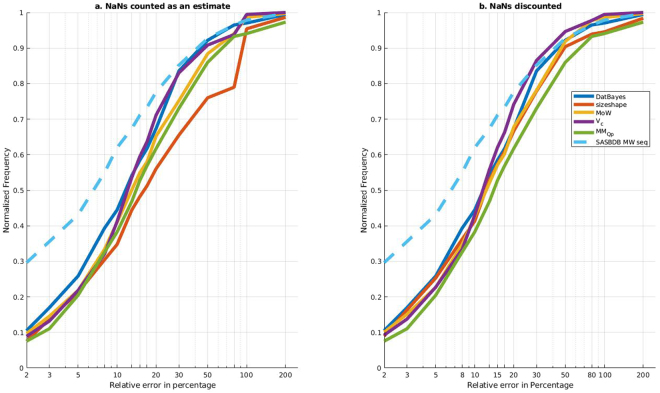
Molecular mass (MM) is one of the key structural parameters obtained by small-angle X-ray scattering (SAXS) of proteins in solution and is used to assess the sample quality, oligomeric composition and to guide subsequent structural modelling. Concentration-dependent assessment of MM relies on a number of extra quantities (partial specific volume, calibrated intensity, accurate solute concentration) and often yields limited accuracy. Concentration-independent methods forgo these requirements being based on the relationship between structural parameters, scattering invariants and particle volume obtained directly from the data. Using a comparative analysis on 165,982 unique scattering profiles calculated from high-resolution protein structures, the performance of multiple concentration-independent MM determination methods was assessed. A Bayesian inference approach was developed affording an accuracy above that of the individual methods, and reports MM estimates together with a credibility interval. This Bayesian approach can be used in combination with concentration-dependent MM methods to further validate the MM of proteins in solution, or as a reliable stand-alone tool in instances where an accurate concentration estimate is not available.
分子量(MM)是通过溶液中蛋白质的小角 X 射线散射(SAXS)获得的关键结构参数之一,用于评估样品质量、寡聚组成,并指导后续的结构建模。基于浓度的 MM 评估依赖于许多额外的量(偏比容、校准强度、准确的溶质浓度),并且通常精度有限。浓度独立的方法无需这些要求,而是基于从数据中直接获得的结构参数、散射不变量和颗粒体积之间的关系。使用对 165982 个独特散射谱图的比较分析,评估了多种浓度独立 MM 测定方法的性能。开发了一种贝叶斯推断方法,其精度高于单个方法,并报告 MM 估计值及其置信区间。这种贝叶斯方法可以与基于浓度的 MM 方法结合使用,以进一步验证溶液中蛋白质的 MM,或者在无法准确估计浓度的情况下作为可靠的独立工具。