Animal Breeding and Genomics, Wageningen University and Research, P.O. Box 338, 6700 AH, Wageningen, The Netherlands.
Biometris, Wageningen University and Research, 6700 AA, Wageningen, The Netherlands.
Genet Sel Evol. 2018 May 18;50(1):27. doi: 10.1186/s12711-018-0396-8.
Genomic prediction (GP) across breeds has so far resulted in low accuracies of the predicted genomic breeding values. Our objective was to evaluate whether using whole-genome sequence (WGS) instead of low-density markers can improve GP across breeds, especially when markers are pre-selected from a genome-wide association study (GWAS), and to test our hypothesis that many non-causal markers in WGS data have a diluting effect on accuracy of across-breed prediction.
Estimated breeding values for stature and bovine high-density (HD) genotypes were available for 595 Jersey bulls from New Zealand, 957 Holstein bulls from New Zealand and 5553 Holstein bulls from the Netherlands. BovineHD genotypes for all bulls were imputed to WGS using Beagle4 and Minimac2. Genomic prediction across the three populations was performed with ASReml4, with each population used as single reference and as single validation sets. In addition to the 50k, HD and WGS, markers that were significantly associated with stature in a large meta-GWAS analysis were selected and used for prediction, resulting in 10 prediction scenarios. Furthermore, we estimated the proportion of genetic variance captured by markers in each scenario.
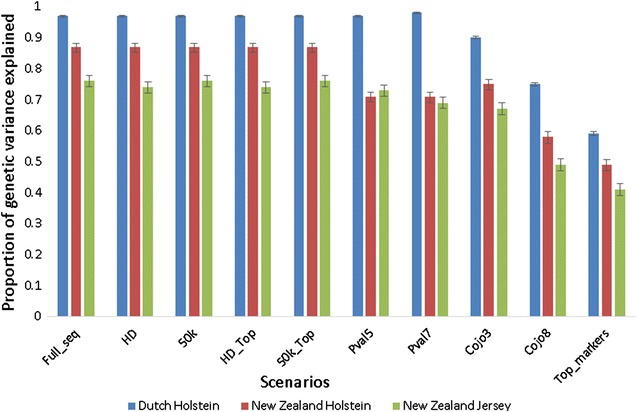
Across breeds, 50k, HD and WGS markers resulted in very low accuracies of prediction ranging from - 0.04 to 0.13. Accuracies were higher in scenarios with pre-selected markers from a meta-GWAS. For example, using only the 133 most significant markers in 133 QTL regions from the meta-GWAS yielded accuracies ranging from 0.08 to 0.23, while 23,125 markers with a - log10(p) higher than 7 resulted in accuracies of up 0.35. Using WGS data did not significantly improve the proportion of genetic variance captured across breeds compared to scenarios with few but pre-selected markers.
Our results demonstrated that the accuracy of across-breed GP can be improved by using markers that are pre-selected from WGS based on their potential causal effect. We also showed that simply increasing the number of markers up to the WGS level does not increase the accuracy of across-breed prediction, even when markers that are expected to have a causal effect are included.
跨品种的基因组预测(GP)迄今为止导致预测基因组育种值的准确性较低。我们的目的是评估使用全基因组序列(WGS)代替低密度标记是否可以提高跨品种的 GP,特别是当标记是从全基因组关联研究(GWAS)中预先选择时,并检验我们的假设,即 WGS 数据中的许多非因果标记对跨品种预测的准确性有稀释作用。
为来自新西兰的 595 头泽西公牛、来自新西兰的 957 头荷斯坦公牛和来自荷兰的 5553 头荷斯坦公牛提供了体型和牛高密度(HD)基因型的估计育种值。使用 Beagle4 和 Minimac2 将所有公牛的 BovineHD 基因型导入 WGS。使用 ASReml4 在三个群体中进行基因组预测,每个群体均用作单一参考和单一验证集。除了 50k、HD 和 WGS 外,还从大型元 GWAS 分析中选择与体型显著相关的标记并用于预测,共产生了 10 种预测方案。此外,我们还估计了每种方案中标记捕获的遗传方差比例。
跨品种,50k、HD 和 WGS 标记的预测准确性非常低,范围从-0.04 到 0.13。在来自元 GWAS 的预先选择标记的方案中,准确性更高。例如,仅使用元 GWAS 中 133 个 QTL 区域的 133 个最显著标记,可产生 0.08 到 0.23 的准确性,而 -log10(p) 值高于 7 的 23,125 个标记则可产生高达 0.35 的准确性。与使用少量但预先选择的标记的方案相比,使用 WGS 数据并不能显著提高跨品种遗传方差的捕获比例。
我们的结果表明,通过使用基于潜在因果效应从 WGS 中预先选择的标记,可以提高跨品种 GP 的准确性。我们还表明,仅增加标记数量到 WGS 水平并不会增加跨品种预测的准确性,即使包含了预期具有因果效应的标记。