Center for Sickle Cell Disease, Howard University, 1840 7th Street, N.W. HURB1, Suite 202, Washington, DC, 20001, USA.
Department of Medicine, Howard University, Washington, DC, USA.
Retrovirology. 2018 May 23;15(1):39. doi: 10.1186/s12977-018-0422-5.
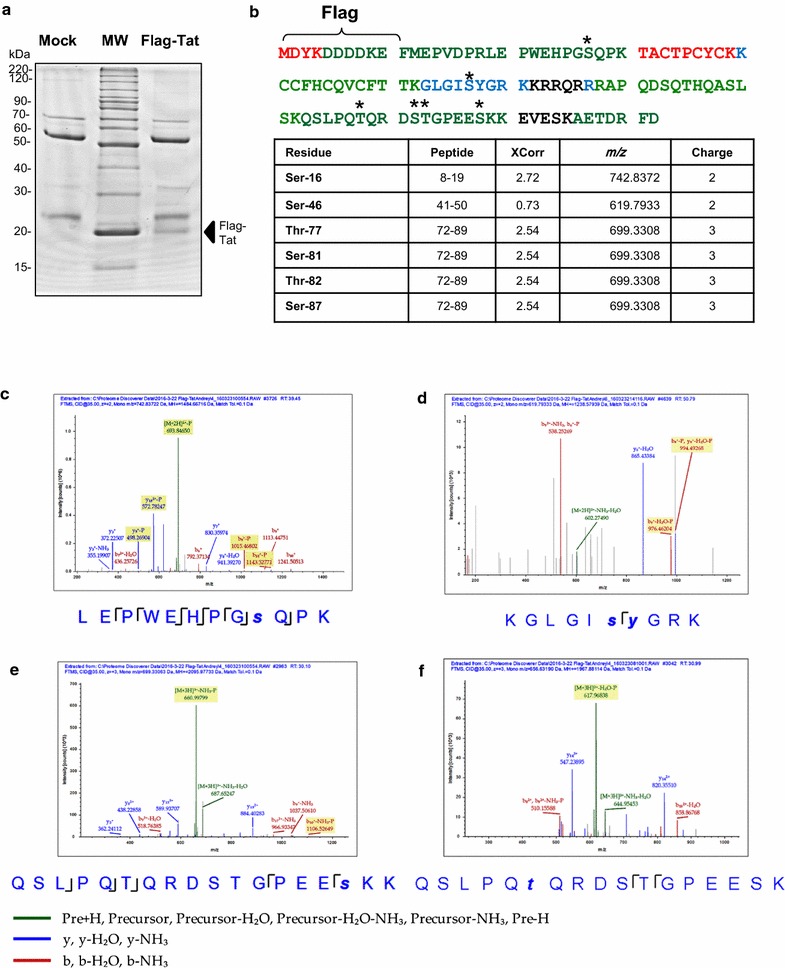
HIV-1 transcription activator protein Tat is phosphorylated in vitro by CDK2 and DNA-PK on Ser-16 residue and by PKR on Tat Ser-46 residue. Here we analyzed Tat phosphorylation in cultured cells and its functionality.
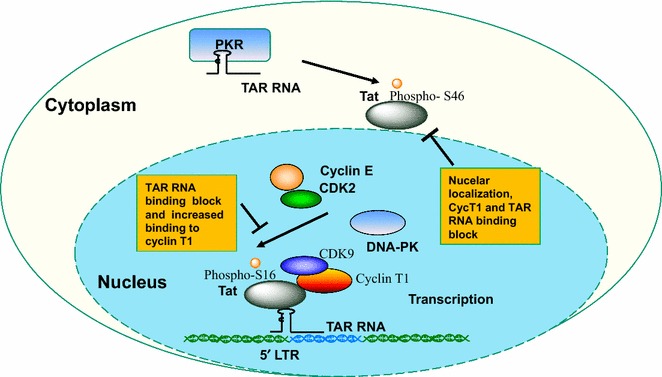
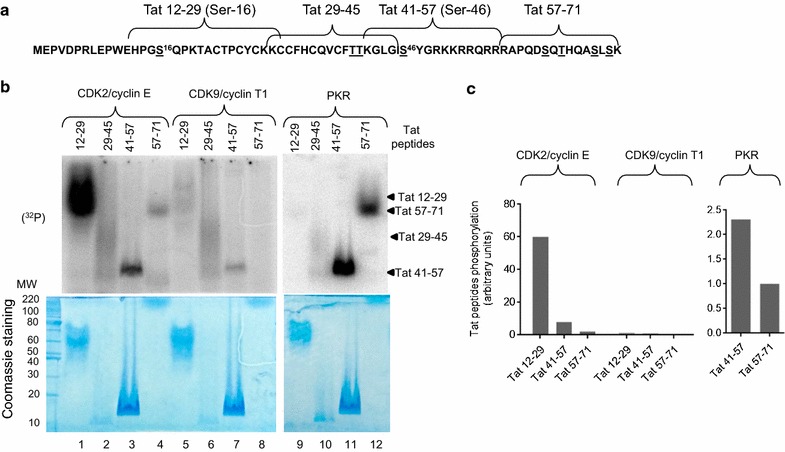
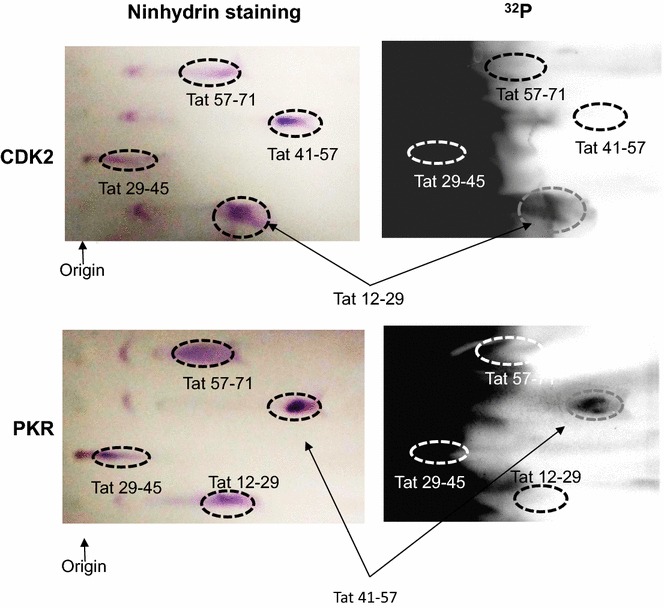
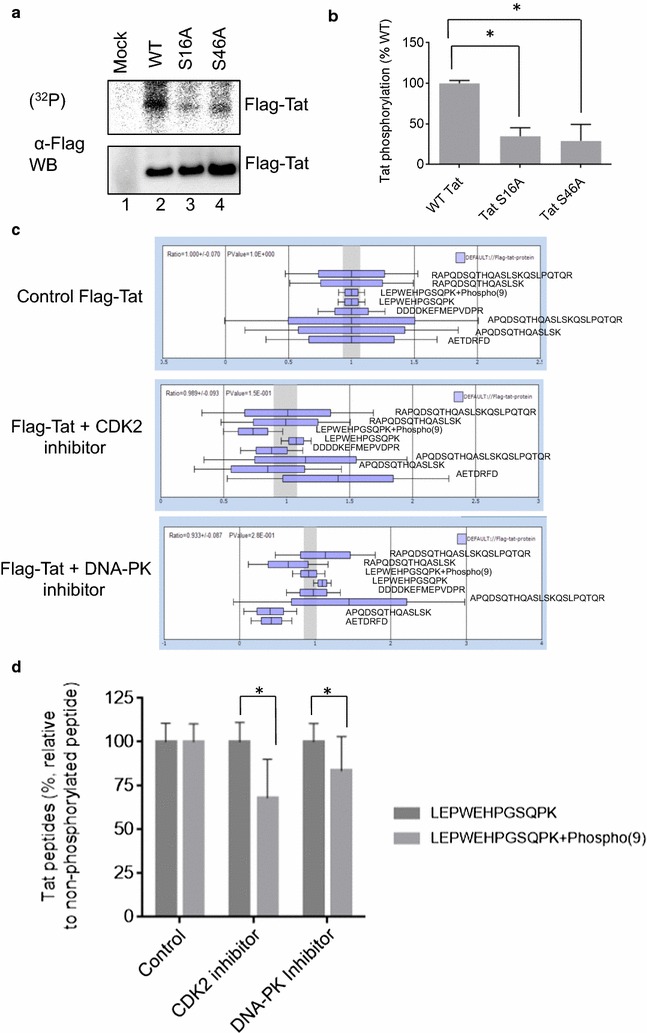
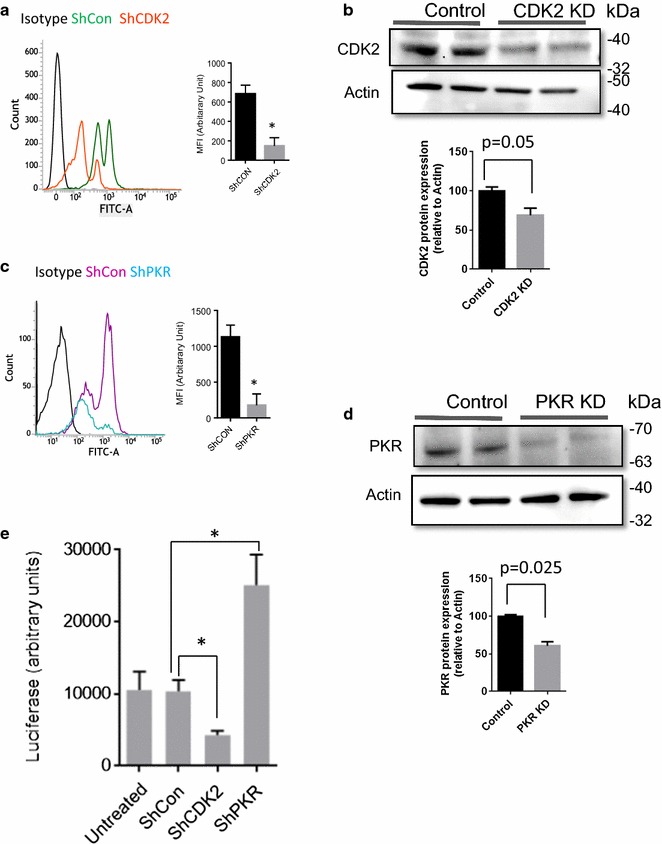
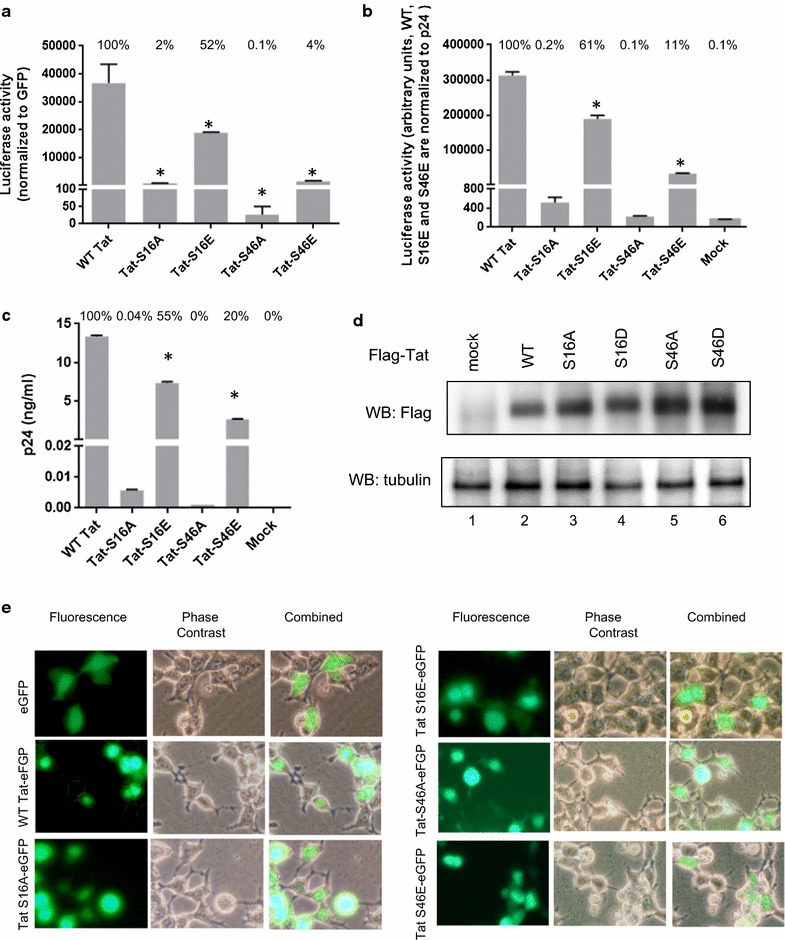
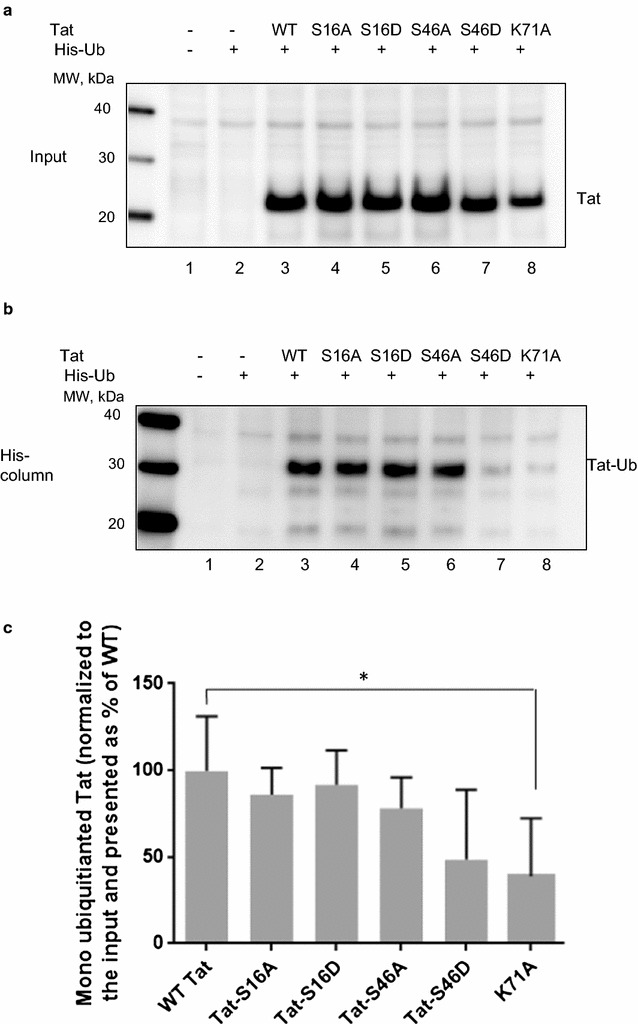
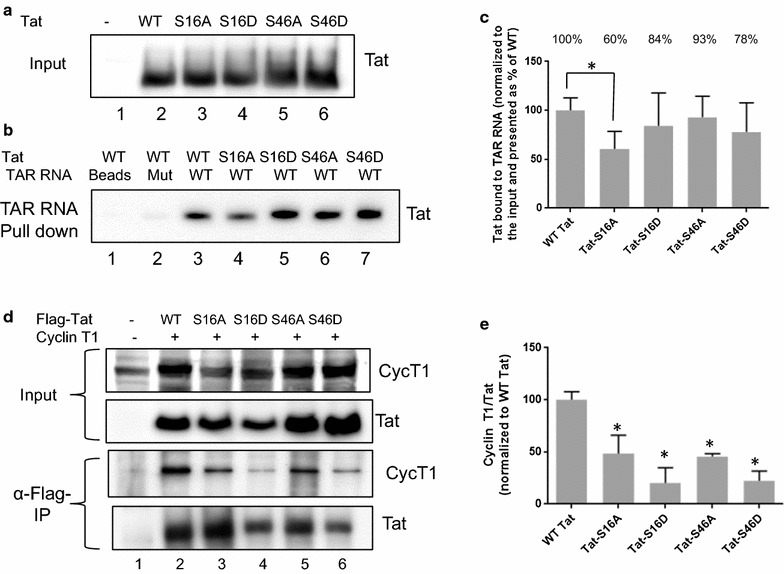
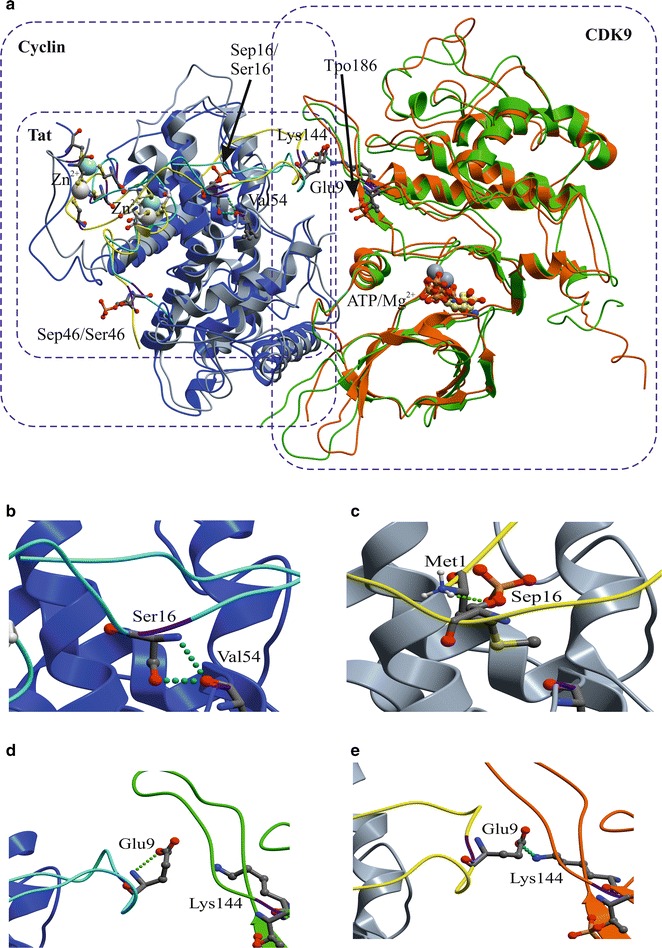
Mass spectrometry analysis showed primarily Tat Ser-16 phosphorylation in cultured cells. In vitro, CDK2/cyclin E predominantly phosphorylated Tat Ser-16 and PKR-Tat Ser-46. Alanine mutations of either Ser-16 or Ser-46 decreased overall Tat phosphorylation. Phosphorylation of Tat Ser-16 was reduced in cultured cells treated by a small molecule inhibitor of CDK2 and, to a lesser extent, an inhibitor of DNA-PK. Conditional knock-downs of CDK2 and PKR inhibited and induced one round HIV-1 replication respectively. HIV-1 proviral transcription was inhibited by Tat alanine mutants and partially restored by S16E mutation. Pseudotyped HIV-1 with Tat S16E mutation replicated well, and HIV-1 Tat S46E-poorly, but no live viruses were obtained with Tat S16A or Tat S46A mutations. TAR RNA binding was affected by Tat Ser-16 alanine mutation. Binding to cyclin T1 showed decreased binding of all Ser-16 and Ser-46 Tat mutants with S16D and Tat S46D mutationts showing the strongest effect. Molecular modelling and molecular dynamic analysis revealed significant structural changes in Tat/CDK9/cyclin T1 complex with phosphorylated Ser-16 residue, but not with phosphorylated Ser-46 residue.
Phosphorylation of Tat Ser-16 induces HIV-1 transcription, facilitates binding to TAR RNA and rearranges CDK9/cyclin T1/Tat complex. Thus, phosphorylation of Tat Ser-16 regulates HIV-1 transcription and may serve as target for HIV-1 therapeutics.
HIV-1 转录激活蛋白 Tat 在体外可被 CDK2 和 DNA-PK 在 Ser-16 残基上磷酸化,也可被 PKR 在 Tat Ser-46 残基上磷酸化。在此,我们分析了培养细胞中的 Tat 磷酸化及其功能。
质谱分析显示,培养细胞中的 Tat 主要磷酸化于 Ser-16 残基。体外实验中,CDK2/细胞周期蛋白 E 优先磷酸化 Tat Ser-16,而 PKR 则磷酸化 Tat Ser-46。Ser-16 或 Ser-46 的丙氨酸突变均降低了 Tat 的整体磷酸化。用 CDK2 的小分子抑制剂处理培养细胞后,Tat Ser-16 的磷酸化减少,而用 DNA-PK 抑制剂处理时则减少较少。CDK2 和 PKR 的条件性敲低分别抑制和诱导一轮 HIV-1 复制。Tat 丙氨酸突变体抑制 HIV-1 前病毒转录,S16E 突变则部分恢复转录。具有 Tat S16E 突变的假型 HIV-1 复制良好,而具有 Tat S46E 突变的 HIV-1 复制较差,但 Tat S16A 或 Tat S46A 突变均未获得活病毒。TAR RNA 结合受 Tat Ser-16 丙氨酸突变的影响。与 cyclin T1 的结合显示,所有 Ser-16 和 Ser-46 Tat 突变体的结合均减弱,而 Tat S46D 突变体的结合则最强。分子建模和分子动力学分析表明,磷酸化 Ser-16 残基而非磷酸化 Ser-46 残基的 Tat/CDK9/cyclin T1 复合物发生了显著的结构变化。
Tat Ser-16 的磷酸化诱导 HIV-1 转录,促进与 TAR RNA 的结合,并重新排列 CDK9/cyclin T1/Tat 复合物。因此,Tat Ser-16 的磷酸化调节 HIV-1 转录,可作为 HIV-1 治疗的靶点。