Key Laboratory of Molecular Cardiovascular Science, Ministry of Education, Beijing, China.
Cell Death Dis. 2018 May 23;9(6):612. doi: 10.1038/s41419-018-0598-6.
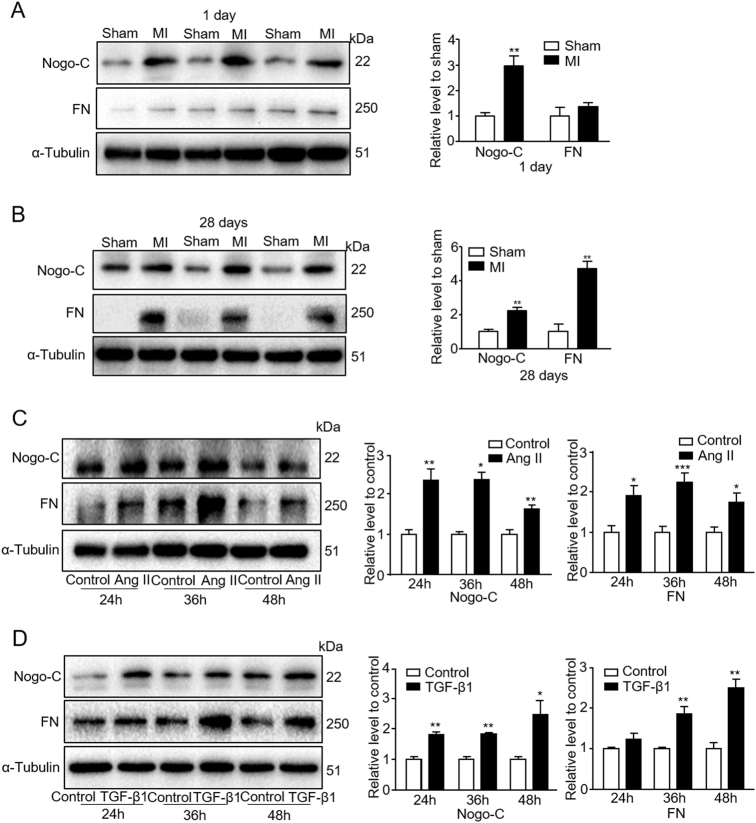
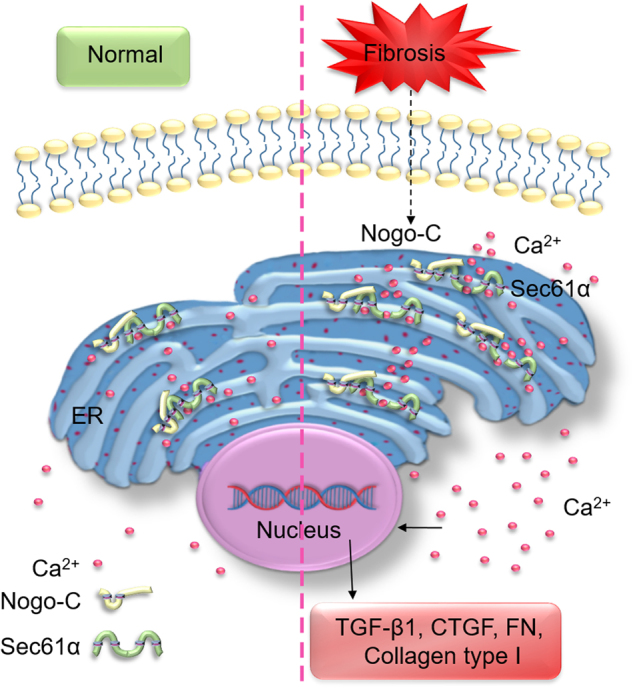
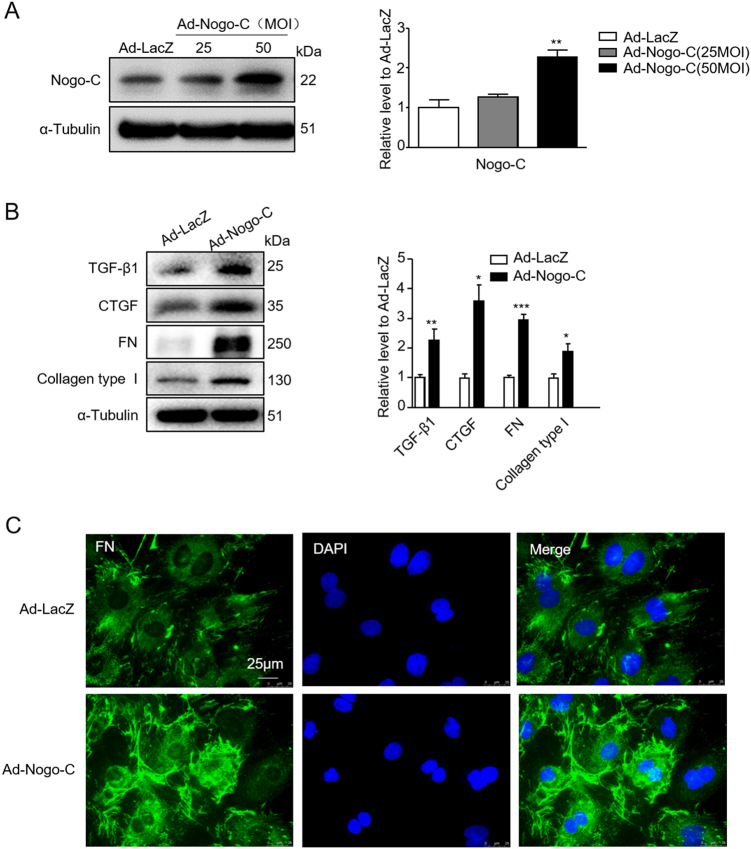
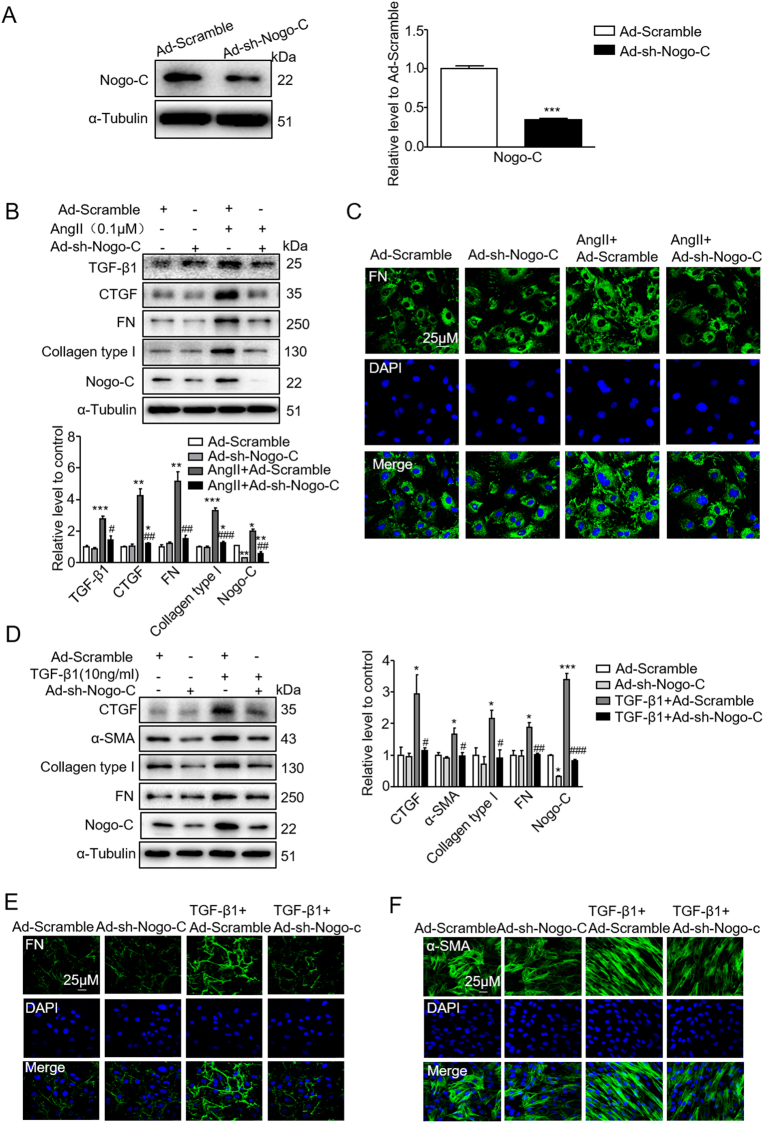
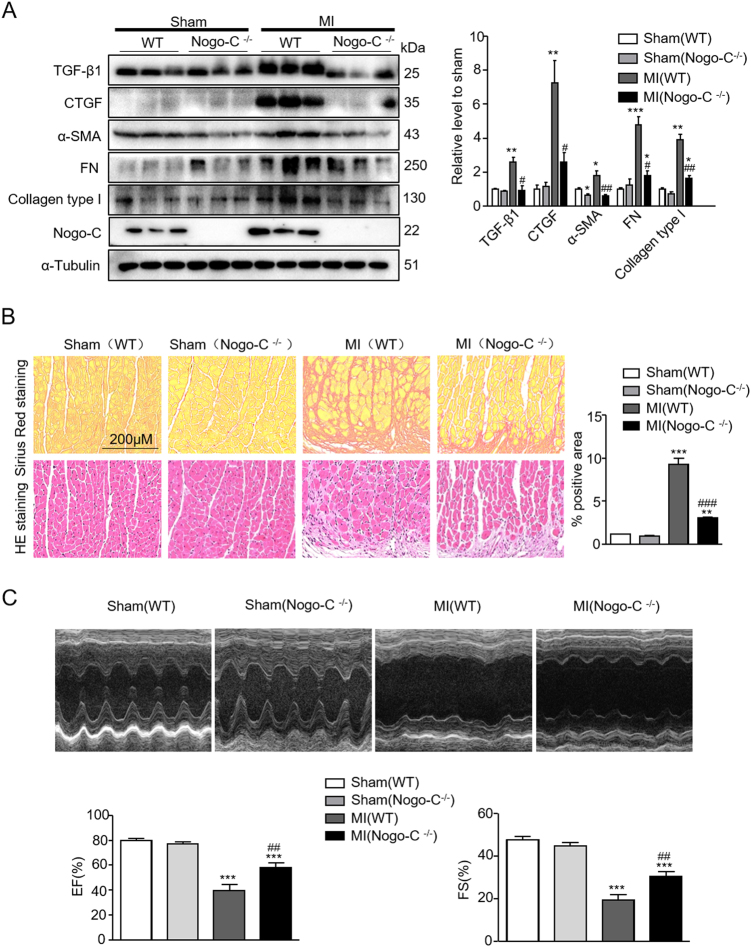
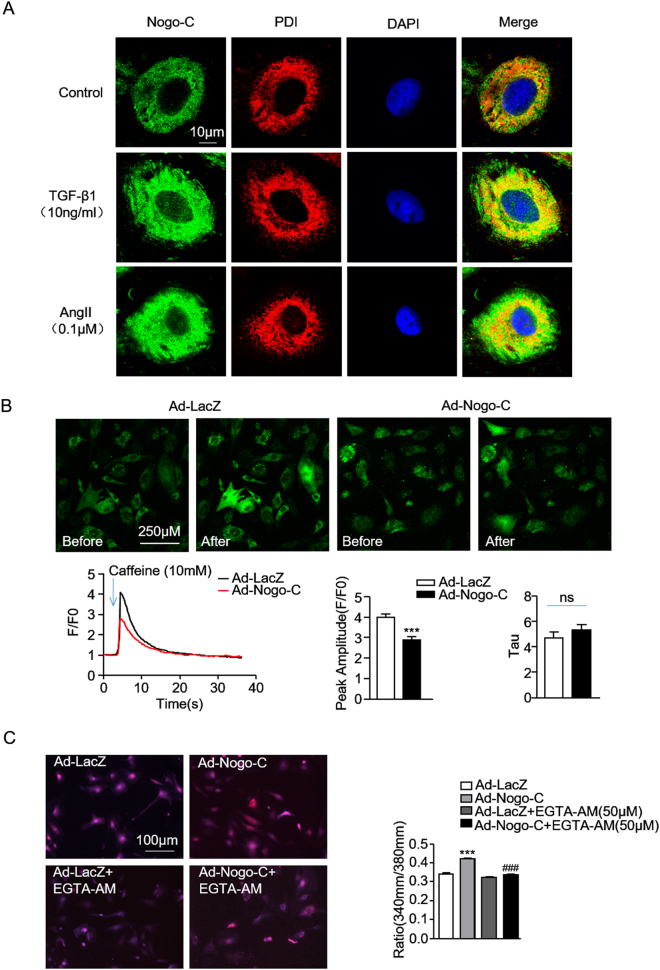
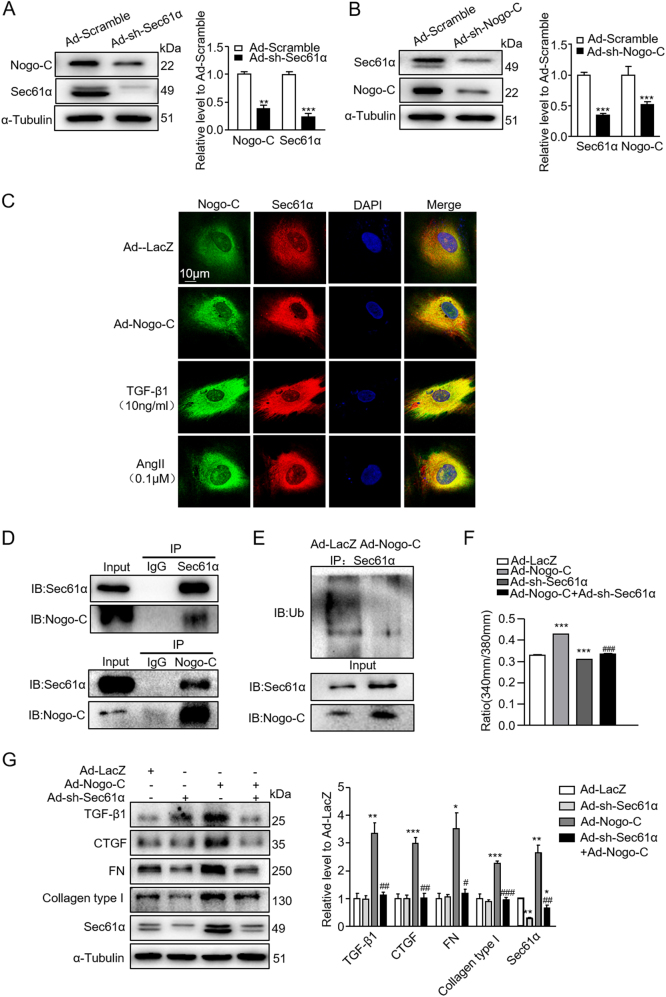
Cardiac fibrosis is an independent risk factor for heart failure and even the leading cause of death in myocardial infarction patients. However, molecular mechanisms associated with the pathogenesis of cardiac fibrosis following myocardial infarction are not yet fully understood. Nogo-C protein ubiquitously expresses in tissues including in the heart. Our previous study found that Nogo-C regulated cardiomyocyte apoptosis during myocardial infarction. In the present study, we found that Nogo-C was upregulated in fibrotic hearts after myocardial infarction and in Ang II- or TGF-β1-stimulated cardiac fibroblasts. Overexpression of Nogo-C in cardiac fibroblasts increased expression of pro-fibrogenic proteins, while knockdown of Nogo-C inhibited the fibrotic responses of cardiac fibroblasts to Ang II- or TGF-β1 stimulation. Functionally, Nogo-C deficiency suppressed pro-fibrogenic proteins in post-myocardial infarction hearts and ameliorated post-myocardial infarction cardiac function. Mechanistically, we found that Nogo-C increased intracellular Ca concentration and buffering Ca totally abolished Nogo-C-induced fibrotic responses. Moreover, overexpression of Nogo-C caused increased Sec61α, the Ca leakage channel on endoplasmic reticulum membrane. Nogo-C interacted with Sec61α on endoplasmic reticulum and stabilized Sec61α protein by inhibiting its ubiquitination. Inhibition or knockdown of Sec61α blocked Nogo-C-induced increase of cytosolic Ca concentration and inhibited Nogo-C- and TGF-β1-induced fibrotic responses in cardiac fibroblasts, suggesting that Nogo-C regulates cardiac fibrosis through interacting with Sec61α to mediate the Ca leakage from endoplasmic reticulum. Thus, our results reveal a novel mechanism underlying cardiac fibrosis following myocardial infarction, and provide a therapeutic strategy for cardiac remodeling related heart diseases.
心肌纤维化是心力衰竭的独立危险因素,甚至是心肌梗死患者死亡的主要原因。然而,心肌梗死后与心肌纤维化发病机制相关的分子机制尚不完全清楚。Nogo-C 蛋白在包括心脏在内的组织中广泛表达。我们之前的研究发现,Nogo-C 在心肌梗死后调节心肌细胞凋亡。在本研究中,我们发现 Nogo-C 在心肌梗死后纤维化心脏和 Ang II 或 TGF-β1 刺激的心肌成纤维细胞中上调。心肌成纤维细胞中 Nogo-C 的过表达增加了促纤维化蛋白的表达,而 Nogo-C 的敲低抑制了 Ang II 或 TGF-β1 刺激对心肌成纤维细胞的纤维化反应。功能上,Nogo-C 缺乏抑制心肌梗死后心脏中的促纤维化蛋白并改善心肌梗死后的心脏功能。在机制上,我们发现 Nogo-C 增加了细胞内 Ca 浓度,并且完全缓冲 Ca 消除了 Nogo-C 诱导的纤维化反应。此外,Nogo-C 的过表达导致内质网膜上的 Ca 泄漏通道 Sec61α增加。Nogo-C 在内质网上与 Sec61α相互作用,并通过抑制其泛素化来稳定 Sec61α 蛋白。Sec61α 的抑制或敲低阻断了 Nogo-C 诱导的胞质 Ca 浓度增加,并抑制了 Nogo-C 和 TGF-β1 在心肌成纤维细胞中诱导的纤维化反应,表明 Nogo-C 通过与 Sec61α 相互作用来调节内质网 Ca 泄漏,从而调节心肌纤维化。因此,我们的结果揭示了心肌梗死后心肌纤维化的新机制,并为与心脏重塑相关的心脏疾病提供了治疗策略。