Stein Catherine M, Sausville Lindsay, Wejse Christian, Sobota Rafal S, Zetola Nicola M, Hill Philip C, Boom W Henry, Scott William K, Sirugo Giorgio, Williams Scott M
Department of Population and Quantitative Health Sciences, Cleveland, OH.
Tuberculosis Research Unit, Case Western Reserve University, Cleveland, OH.
Curr Genet Med Rep. 2017 Dec;5(4):149-166. doi: 10.1007/s40142-017-0130-9. Epub 2017 Oct 12.
Tuberculosis (TB), caused by (MTB), remains a major public health threat globally. Several lines of evidence support a role for host genetic factors in resistance/susceptibility to TB disease and MTB infection. However, results across candidate gene and genome-wide association studies (GWAS) are largely inconsistent, so a cohesive genetic model underlying TB risk has not emerged.
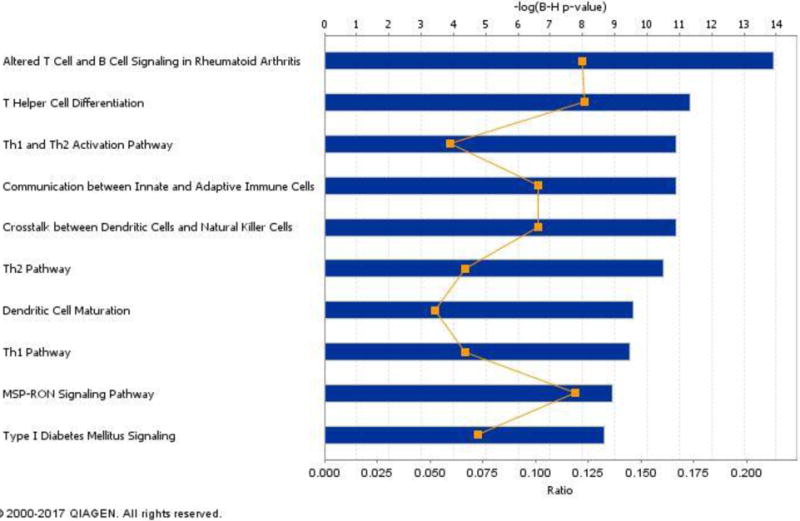
Despite the difficulties in identifying consistent genetic associations, genetic studies of TB and MTB infection have revealed a few well-documented loci. These well validated genes are presented in this review, but there remains a large gap in how these genes translate into better understanding of TB. To address this, we present a pathway based extension of standard association analyses, seeding the results with the best validated genes from candidate gene and GWAS studies.
Several pathways were significantly enriched using pathway analyses that may help to explain population patterns of TB risk. In conclusion, we advocate for novel approaches to the study of host genetic analysis of TB that extend traditional association approaches.
由结核分枝杆菌(MTB)引起的结核病(TB)仍是全球主要的公共卫生威胁。多项证据支持宿主遗传因素在结核病发病和MTB感染的抗性/易感性中起作用。然而,候选基因研究和全基因组关联研究(GWAS)的结果在很大程度上并不一致,因此尚未形成一个连贯的结核病风险遗传模型。
尽管在确定一致的遗传关联方面存在困难,但结核病和MTB感染的遗传学研究已经揭示了一些有充分文献记载的基因座。本综述介绍了这些经过充分验证的基因,但在如何将这些基因转化为对结核病的更好理解方面仍存在很大差距。为了解决这个问题,我们提出了一种基于通路的标准关联分析扩展方法,将候选基因研究和GWAS研究中经过最佳验证的基因作为结果的起始点。
使用通路分析发现有几个通路显著富集,这可能有助于解释结核病风险的人群模式。总之,我们提倡采用新方法来研究结核病的宿主遗传分析,扩展传统的关联方法。