Shulskaya Marina V, Alieva Anelya Kh, Vlasov Ivan N, Zyrin Vladimir V, Fedotova Ekaterina Yu, Abramycheva Natalia Yu, Usenko Tatiana S, Yakimovsky Andrei F, Emelyanov Anton K, Pchelina Sofya N, Illarioshkin Sergei N, Slominsky Petr A, Shadrina Maria I
Laboratory of Molecular Genetics of Hereditary Diseases, Institute of Molecular Genetics, Russian Academy of Sciences (RAS), Moscow, Russia.
Federal State Scientific Institution, Scientific Center of Neurology, Russian Academy of Sciences (RAS), Moscow, Russia.
Front Aging Neurosci. 2018 May 15;10:136. doi: 10.3389/fnagi.2018.00136. eCollection 2018.
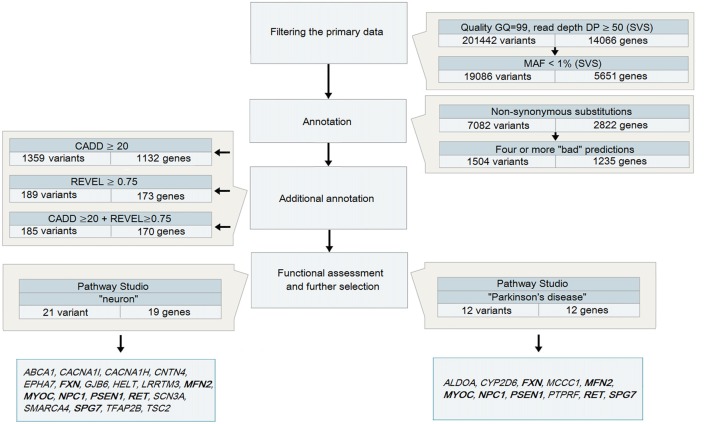
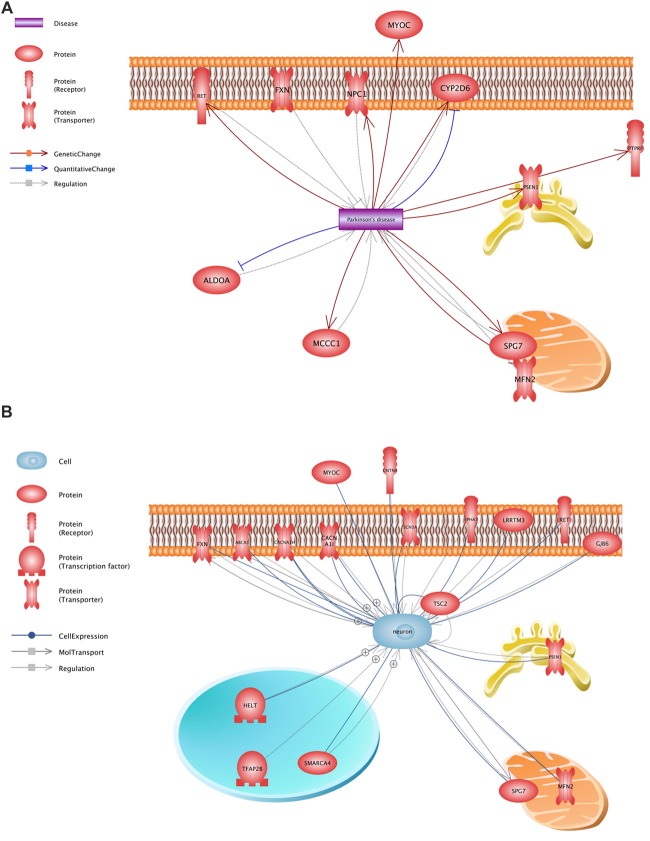
: Parkinson's disease (PD) is a complex disease with its monogenic forms accounting for less than 10% of all cases. Whole-exome sequencing (WES) technology has been used successfully to find mutations in large families. However, because of the late onset of the disease, only small families and unrelated patients are usually available. WES conducted in such cases yields in a large number of candidate variants. There are currently a number of imperfect software tools that allow the pathogenicity of variants to be evaluated. : We analyzed 48 unrelated patients with an alleged autosomal dominant familial form of PD using WES and developed a strategy for selecting potential pathogenetically significant variants using almost all available bioinformatics resources for the analysis of exonic areas. : DNA sequencing of 48 patients with excluded frequent mutations was performed using an Illumina HiSeq 2500 platform. The possible pathogenetic significance of identified variants and their involvement in the pathogenesis of PD was assessed using SNP and Variation Suite (SVS), Combined Annotation Dependent Depletion (CADD) and Rare Exome Variant Ensemble Learner (REVEL) software. Functional evaluation was performed using the Pathway Studio database. : A significant reduction in the search range from 7082 to 25 variants in 23 genes associated with PD or neuronal function was achieved. Eight (, , , , , , and ) were the most significant. : The multistep approach developed made it possible to conduct an effective search for potential pathogenetically significant variants, presumably involved in the pathogenesis of PD. The data obtained need to be further verified experimentally.
帕金森病(PD)是一种复杂疾病,其单基因形式在所有病例中占比不到10%。全外显子组测序(WES)技术已成功用于在大家族中寻找突变。然而,由于该疾病发病较晚,通常只有小家族和无血缘关系的患者可供研究。在此类病例中进行的WES会产生大量候选变异。目前有一些不完善的软件工具可用于评估变异的致病性。我们使用WES分析了48例据称患有常染色体显性遗传家族性帕金森病的无血缘关系患者,并开发了一种策略,利用几乎所有可用的生物信息学资源来分析外显子区域,以选择具有潜在致病意义的变异。使用Illumina HiSeq 2500平台对48例排除常见突变的患者进行DNA测序。使用SNP和变异套件(SVS)、联合注释依赖损耗(CADD)和罕见外显子变异集合学习器(REVEL)软件评估已识别变异的可能致病意义及其在帕金森病发病机制中的作用。使用Pathway Studio数据库进行功能评估。在与帕金森病或神经元功能相关的23个基因中,搜索范围从7082个显著减少到25个变异。其中8个( 、 、 、 、 、 、 和 )最为显著。所开发的多步骤方法使得能够有效搜索可能参与帕金森病发病机制的潜在致病意义变异。所获得的数据需要进一步通过实验进行验证。