Li Ginny X H, Vogel Christine, Choi Hyungwon
Saw Swee Hock School of Public Health, National University of Singapore, Singapore.
Mol Omics. 2018 Jun 12;14(3):197-209. doi: 10.1039/c8mo00027a.
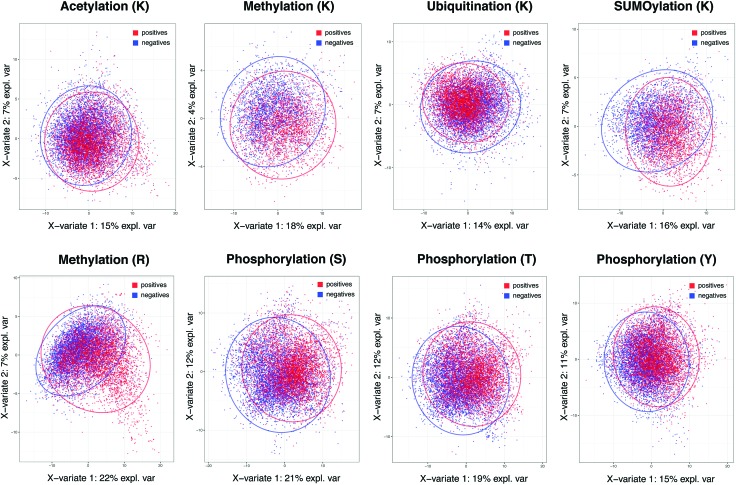
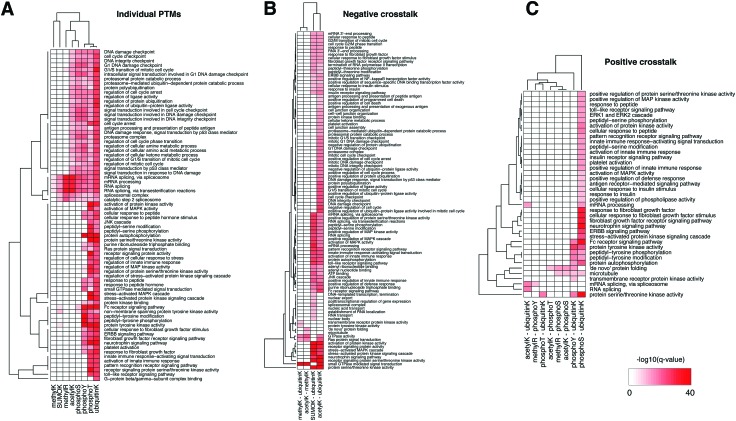
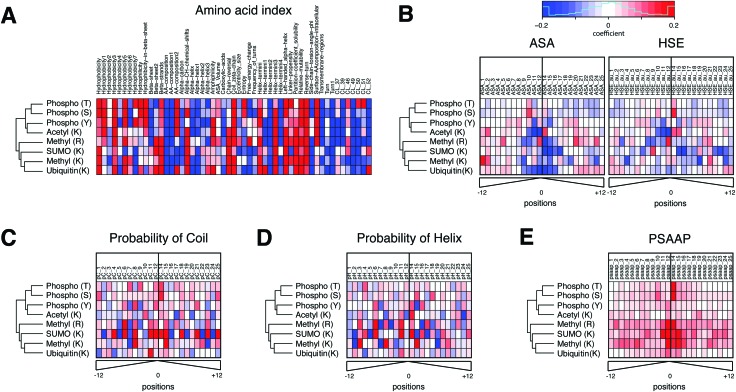
While tandem mass spectrometry can detect post-translational modifications (PTM) at the proteome scale, reported PTM sites are often incomplete and include false positives. Computational approaches can complement these datasets by additional predictions, but most available tools use prediction models pre-trained for single PTM type by the developers and it remains a difficult task to perform large-scale batch prediction for multiple PTMs with flexible user control, including the choice of training data. We developed an R package called PTMscape which predicts PTM sites across the proteome based on a unified and comprehensive set of descriptors of the physico-chemical microenvironment of modified sites, with additional downstream analysis modules to test enrichment of individual or pairs of PTMs in protein domains. PTMscape is flexible in the ability to process any major modifications, such as phosphorylation and ubiquitination, while achieving the sensitivity and specificity comparable to single-PTM methods and outperforming other multi-PTM tools. Applying this framework, we expanded proteome-wide coverage of five major PTMs affecting different residues by prediction, especially for lysine and arginine modifications. Using a combination of experimentally acquired sites (PSP) and newly predicted sites, we discovered that the crosstalk among multiple PTMs occur more frequently than by random chance in key protein domains such as histone, protein kinase, and RNA recognition motifs, spanning various biological processes such as RNA processing, DNA damage response, signal transduction, and regulation of cell cycle. These results provide a proteome-scale analysis of crosstalk among major PTMs and can be easily extended to other types of PTM.
虽然串联质谱可以在蛋白质组规模上检测翻译后修饰(PTM),但已报道的PTM位点往往不完整且包含假阳性。计算方法可以通过额外的预测来补充这些数据集,但大多数现有工具使用开发者预先为单一PTM类型训练的预测模型,对于多种PTM进行大规模批量预测并实现灵活的用户控制(包括训练数据的选择)仍然是一项艰巨的任务。我们开发了一个名为PTMscape的R包,它基于修饰位点物理化学微环境的统一且全面的描述符集预测整个蛋白质组中的PTM位点,并带有额外的下游分析模块来测试蛋白质结构域中单个或成对PTM的富集情况。PTMscape在处理任何主要修饰(如磷酸化和泛素化)方面具有灵活性,同时实现了与单PTM方法相当的灵敏度和特异性,并且优于其他多PTM工具。应用这个框架,我们通过预测扩展了对影响不同残基的五种主要PTM的全蛋白质组覆盖范围,特别是对于赖氨酸和精氨酸修饰。使用实验获得的位点(PSP)和新预测的位点相结合的方法,我们发现多个PTM之间的串扰在关键蛋白质结构域(如组蛋白、蛋白激酶和RNA识别基序)中比随机情况更频繁地发生,这些结构域跨越各种生物学过程,如RNA加工、DNA损伤反应、信号转导和细胞周期调控。这些结果提供了对主要PTM之间串扰的蛋白质组规模分析,并且可以很容易地扩展到其他类型的PTM。