Sainio Markus T, Ylikallio Emil, Mäenpää Laura, Lahtela Jenni, Mattila Pirkko, Auranen Mari, Palmio Johanna, Tyynismaa Henna
Research Programs Unit (M.T.S., E.Y., L.M., M.A., H.T.), Molecular Neurology, University of Helsinki; Clinical Neurosciences, Neurology (E.Y., M.A.), University of Helsinki and Helsinki University Hospital; Institute for Molecular Medicine Finland (FIMM) (J.L., P.M.), University of Helsinki; Neuromuscular Research Center (J.P.), Tampere University Hospital and University of Tampere; and Department of Medical and Clinical Genetics (H.T.), University of Helsinki, Finland.
Neurol Genet. 2018 Jun 5;4(3):e244. doi: 10.1212/NXG.0000000000000244. eCollection 2018 Jun.
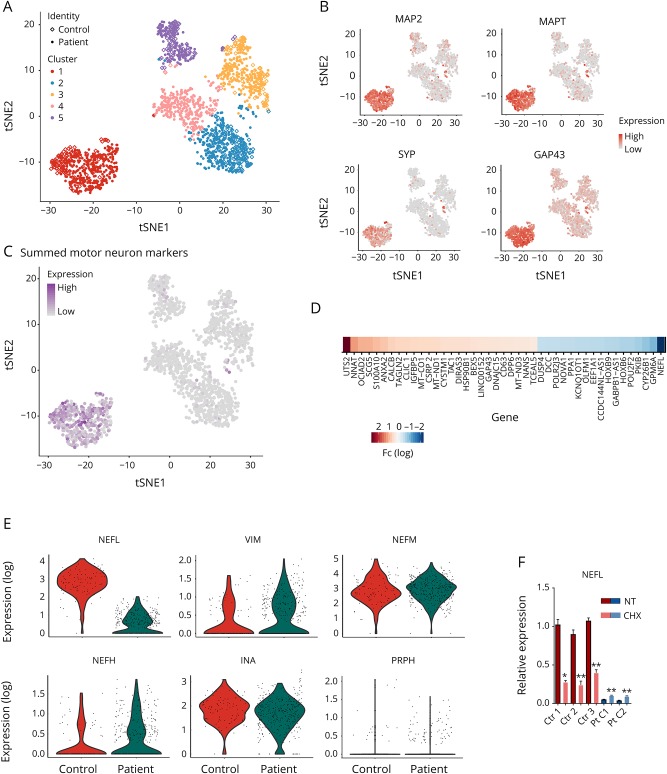
We used patient-specific neuronal cultures to characterize the molecular genetic mechanism of recessive nonsense mutations in neurofilament light () underlying early-onset Charcot-Marie-Tooth (CMT) disease.
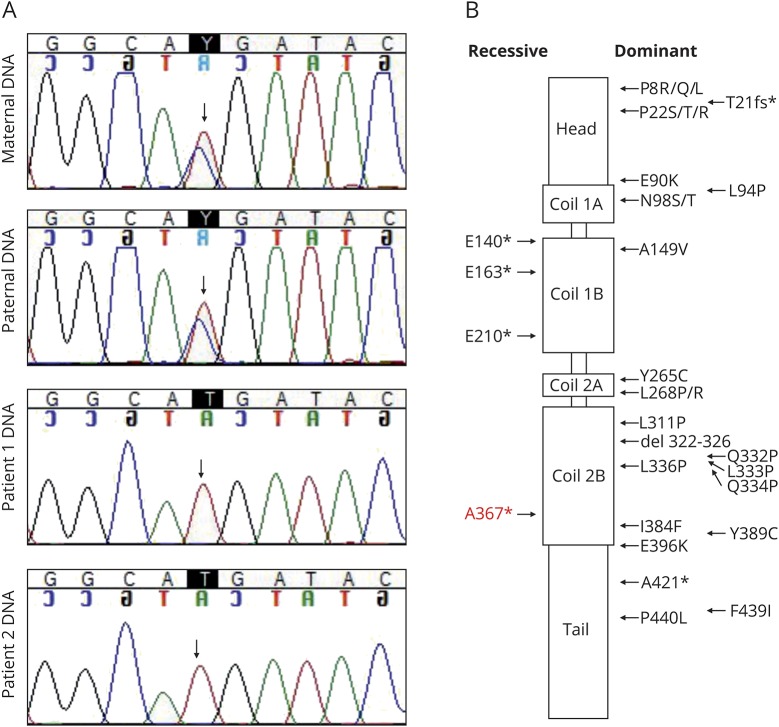
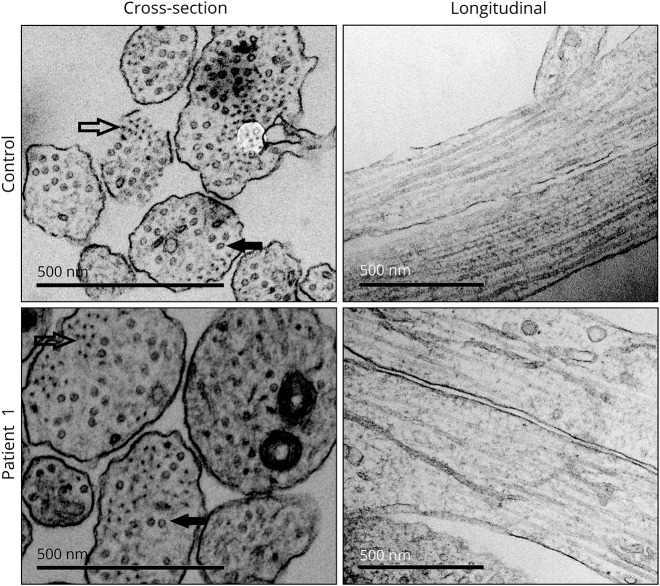
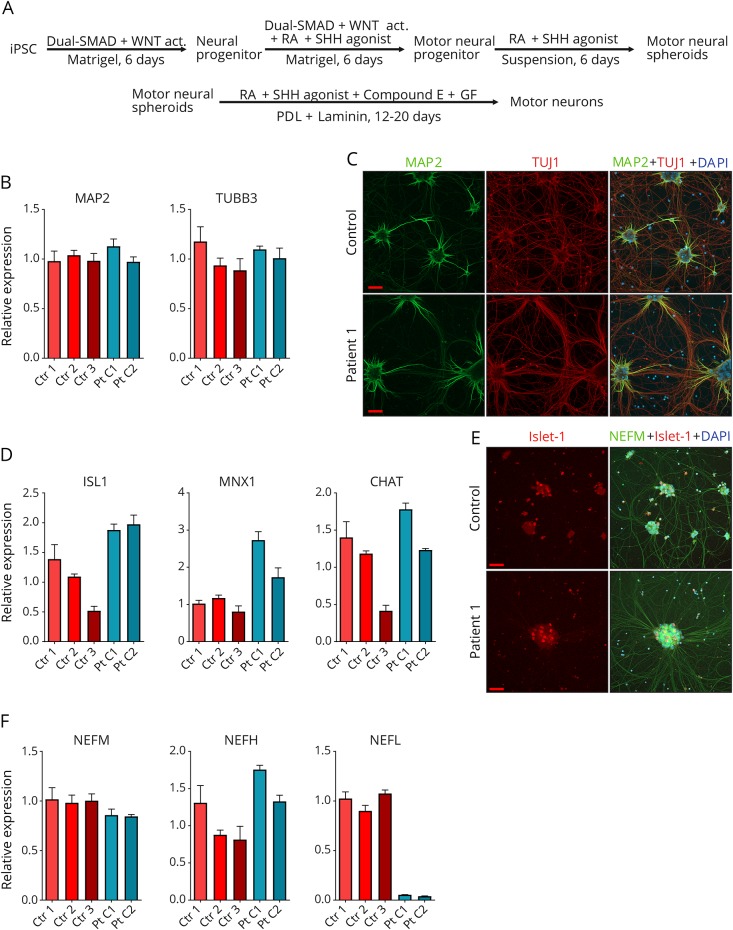
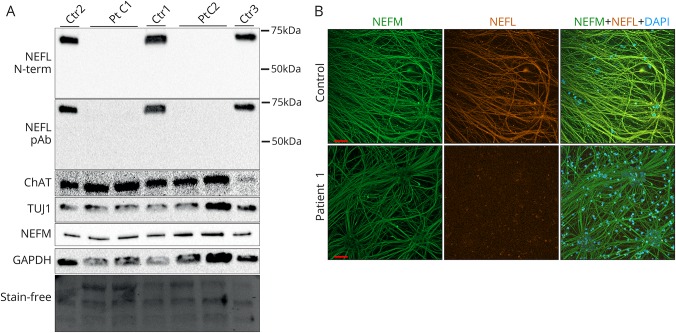
Motor neurons were differentiated from induced pluripotent stem cells of a patient with early-onset CMT carrying a novel homozygous nonsense mutation in . Quantitative PCR, protein analytics, immunocytochemistry, electron microscopy, and single-cell transcriptomics were used to investigate patient and control neurons.
We show that the recessive nonsense mutation causes a nearly total loss of messenger RNA (mRNA), leading to the complete absence of NEFL protein in patient's cultured neurons. Yet the cultured neurons were able to differentiate and form neuronal networks and neurofilaments. Single-neuron gene expression fingerprinting pinpointed as the most downregulated gene in the patient neurons and provided data of intermediate filament transcript abundancy and dynamics in cultured neurons. Blocking of nonsense-mediated decay partially rescued the loss of mRNA.
The strict neuronal specificity of neurofilament has hindered the mechanistic studies of recessive nonsense mutations. Here, we show that such mutation leads to the absence of NEFL, causing childhood-onset neuropathy through a loss-of-function mechanism. We propose that the neurofilament accumulation, a common feature of many neurodegenerative diseases, mimics the absence of NEFL seen in recessive CMT if aggregation prevents the proper localization of wild-type NEFL in neurons. Our results suggest that the removal of NEFL as a proposed treatment option is harmful in humans.
我们使用患者特异性神经元培养物来表征神经丝轻链(NEFL)隐性无义突变在早发性夏科-马里-图斯病(CMT)发病机制中的分子遗传学机制。
运动神经元从一名携带NEFL基因新的纯合无义突变的早发性CMT患者的诱导多能干细胞分化而来。采用定量PCR、蛋白质分析、免疫细胞化学、电子显微镜和单细胞转录组学来研究患者和对照神经元。
我们发现隐性无义突变导致NEFL信使核糖核酸(mRNA)几乎完全缺失,致使患者培养的神经元中完全没有NEFL蛋白。然而,培养的神经元能够分化并形成神经网络和神经丝。单神经元基因表达指纹图谱确定NEFL是患者神经元中下调最明显的基因,并提供了培养神经元中中间丝转录本丰度和动态的数据。阻断无义介导的衰变部分挽救了NEFL mRNA的缺失。
神经丝严格的神经元特异性阻碍了对隐性NEFL无义突变机制的研究。在此,我们表明这种突变导致NEFL缺失,通过功能丧失机制导致儿童期神经病。我们提出,神经丝积聚是许多神经退行性疾病的共同特征,如果聚集阻止野生型NEFL在神经元中的正确定位,那么它会模拟隐性CMT中所见的NEFL缺失。我们的结果表明,去除NEFL作为一种提议的治疗选择对人类有害。