Yale University School of Public Health, New Haven, CT, USA.
Yale University School of Nursing, West Haven, CT, USA.
BMC Infect Dis. 2018 Jun 22;18(1):282. doi: 10.1186/s12879-018-3186-6.
Dengue and West Nile viruses are highly cross-reactive and have numerous parallels in geography, potential vector host (Aedes family of mosquitoes), and initial symptoms of infection. While the vast majority (> 80%) of both dengue and West Nile virus infections result in asymptomatic infections, a minority of individuals experience symptomatic infection and an even smaller proportion develop severe disease. The mechanisms by which these infections lead to severe disease in a subset of infected individuals is incompletely understood, but individual host differences including genetic factors and immune responses have been proposed. We sought to identify genetic risk factors that are associated with more severe disease outcomes for both viruses in order to shed light on possible shared mechanisms of resistance and potential therapeutic interventions.
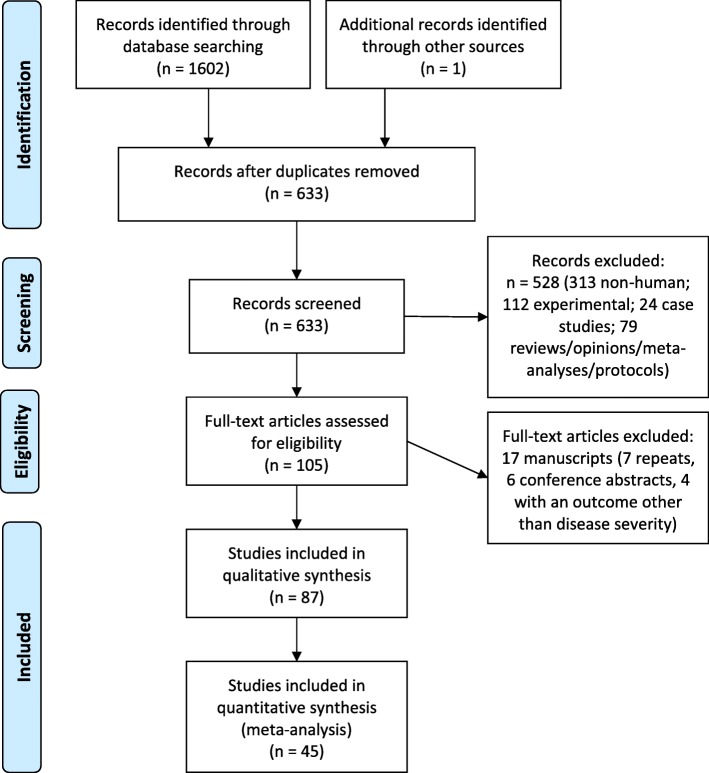
We applied a search strategy using four major databases (Medline, PubMed, Embase, and Global Health) to find all known genetic associations identified to date with dengue or West Nile virus disease. Here we present a review of our findings and a meta-analysis of genetic variants identified.
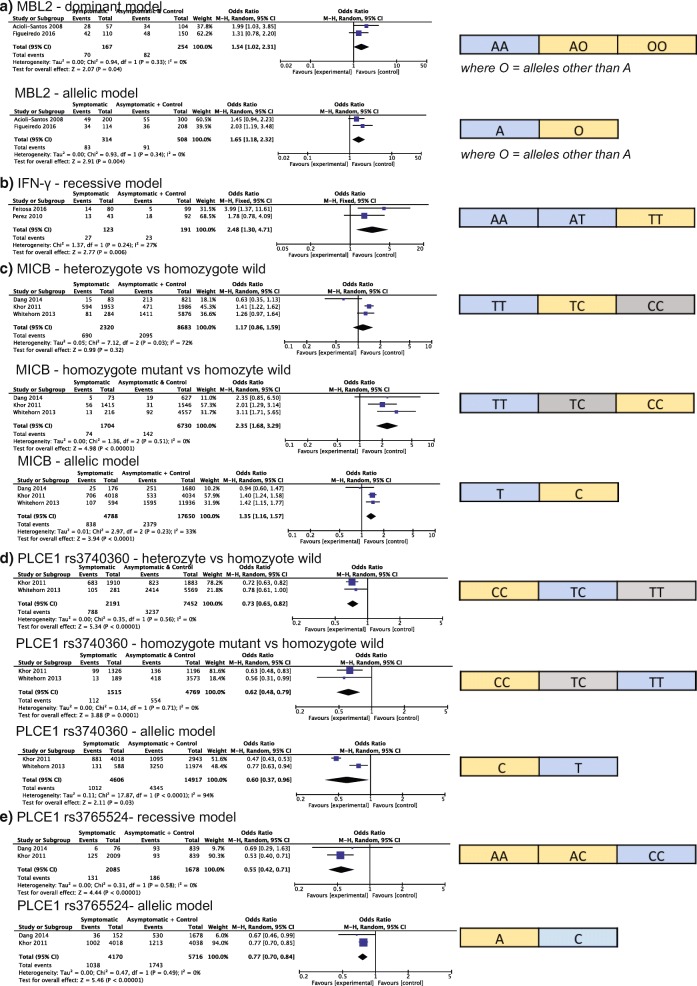
We found genetic variations that are significantly associated with infections of these viruses. In particular we found variation within the OAS1 (meta-OR = 0.83, 95% CI: 0.69-1.00) and CCR5 (meta-OR = 1.29, 95% CI: 1.08-1.53) genes is significantly associated with West Nile virus disease, while variation within MICB (meta-OR = 2.35, 95% CI: 1.68-3.29), PLCE1 (meta-OR = 0.55, 95% CI: 0.42-0.71), MBL2 (meta-OR = 1.54, 95% CI: 1.02-2.31), and IFN-γ (meta-OR = 2.48, 95% CI: 1.30-4.71), is associated with dengue disease.
Despite substantial heterogeneity in populations studied, genes examined, and methodology, significant associations with genetic variants were found across studies within both diseases. These gene associations suggest a key role for immune mechanisms in susceptibility to severe disease. Further research is needed to elucidate the role of these genes in disease pathogenesis and may reveal additional genetic factors associated with disease severity.
登革热和西尼罗河病毒高度交叉反应,在地理、潜在媒介宿主(伊蚊科蚊子)和感染初始症状方面有许多相似之处。虽然登革热和西尼罗河病毒感染的绝大多数(>80%)导致无症状感染,但少数人出现症状性感染,极少数人发展为严重疾病。这些感染导致部分感染者发生严重疾病的机制尚不完全清楚,但已提出个体宿主差异包括遗传因素和免疫反应。我们试图确定与这两种病毒的更严重疾病结果相关的遗传风险因素,以便阐明可能的共同抵抗机制和潜在的治疗干预措施。
我们应用了一种搜索策略,使用四个主要数据库(Medline、PubMed、Embase 和 Global Health)查找迄今为止与登革热或西尼罗河病毒病相关的所有已知遗传关联。在这里,我们介绍了我们的研究结果的综述和遗传变异的荟萃分析。
我们发现了与这些病毒感染显著相关的遗传变异。特别是,我们发现 OAS1(meta-OR=0.83,95%CI:0.69-1.00)和 CCR5(meta-OR=1.29,95%CI:1.08-1.53)基因内的变异与西尼罗河病毒病显著相关,而 MICB(meta-OR=2.35,95%CI:1.68-3.29)、PLCE1(meta-OR=0.55,95%CI:0.42-0.71)、MBL2(meta-OR=1.54,95%CI:1.02-2.31)和 IFN-γ(meta-OR=2.48,95%CI:1.30-4.71)内的变异与登革热疾病相关。
尽管研究人群、检查基因和方法学存在很大异质性,但在这两种疾病的研究中都发现了与遗传变异的显著关联。这些基因关联表明免疫机制在易感性严重疾病中起关键作用。需要进一步研究以阐明这些基因在疾病发病机制中的作用,并可能揭示与疾病严重程度相关的其他遗传因素。