Department of Pharmaceutical Sciences , University of Maryland School of Pharmacy , Baltimore , Maryland 21201 , United States.
J Chem Inf Model. 2018 Jul 23;58(7):1372-1383. doi: 10.1021/acs.jcim.8b00227. Epub 2018 Jul 11.
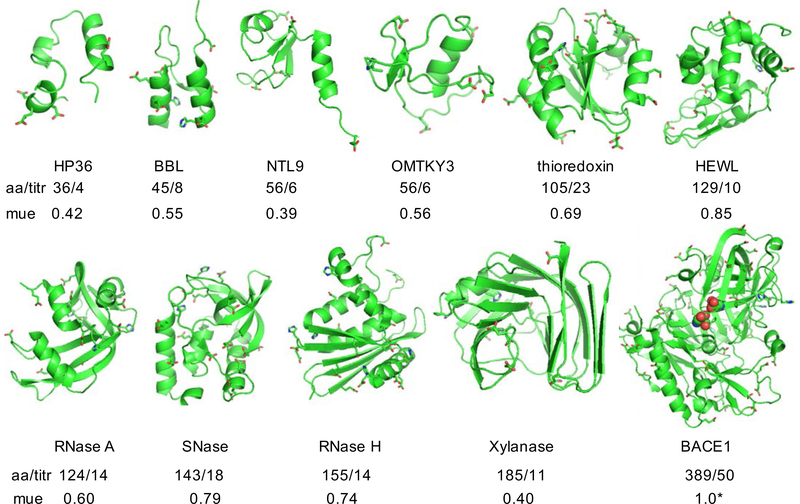
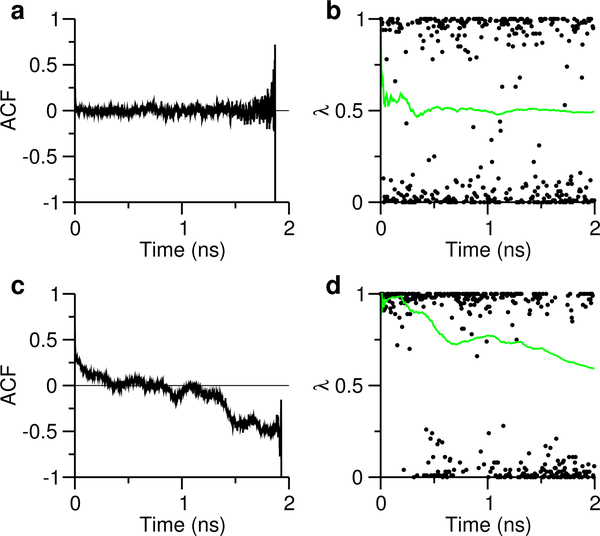
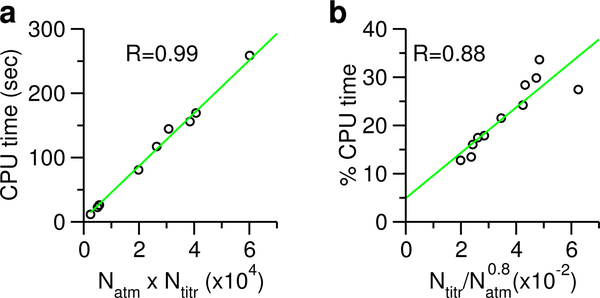
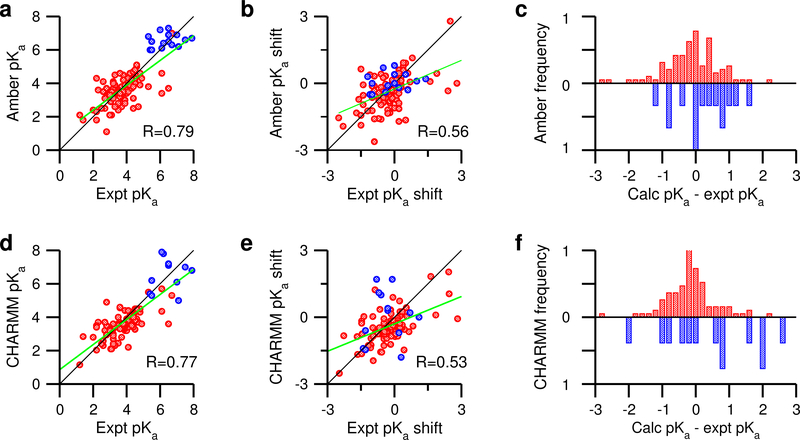
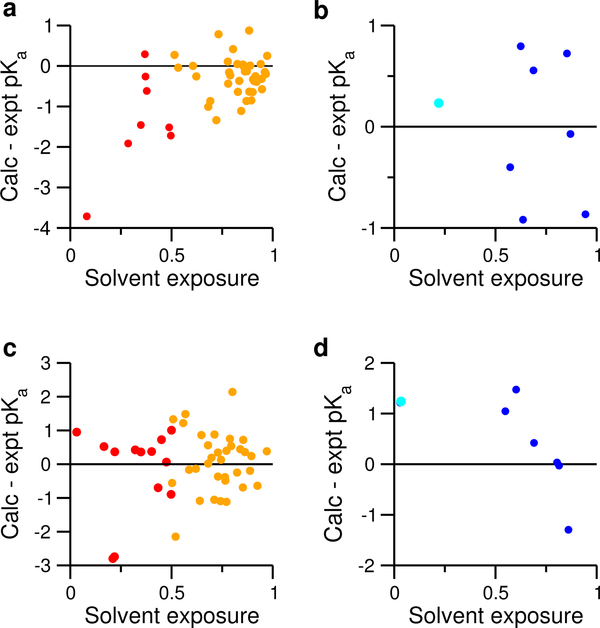
Solution pH plays an important role in structure and dynamics of biomolecular systems; however, pH effects cannot be accurately accounted for in conventional molecular dynamics simulations based on fixed protonation states. Continuous constant pH molecular dynamics (CpHMD) based on the λ-dynamics framework calculates protonation states on the fly during dynamical simulation at a specified pH condition. Here we report the CPU-based implementation of the CpHMD method based on the GBNeck2 generalized Born (GB) implicit-solvent model in the pmemd engine of the Amber molecular dynamics package. The performance of the method was tested using pH replica-exchange titration simulations of Asp, Glu and His side chains in 4 miniproteins and 7 enzymes with experimentally known p K's, some of which are significantly shifted from the model values. The added computational cost due to CpHMD titration ranges from 11 to 33% for the data set and scales roughly linearly as the ratio between the titrable sites and number of solute atoms. Comparison of the experimental and calculated p K's using 2 ns per replica sampling yielded a mean unsigned error of 0.70, a root-mean-squared error of 0.91, and a linear correlation coefficient of 0.79. Though this level of accuracy is similar to the GBSW-based CpHMD in CHARMM, in contrast to the latter, the current implementation was able to reproduce the experimental orders of the p K's of the coupled carboxylic dyads. We quantified the sampling errors, which revealed that prolonged simulation is needed to converge p K's of several titratable groups involved in salt-bridge-like interactions or deeply buried in the protein interior. Our benchmark data demonstrate that GBNeck2-CpHMD is an attractive tool for protein p K predictions.
溶液 pH 值在生物分子体系的结构和动力学中起着重要作用;然而,在基于固定质子化状态的传统分子动力学模拟中,无法准确考虑 pH 值的影响。基于 λ 动力学框架的连续恒 pH 值分子动力学(CpHMD)在指定 pH 值条件下的动力学模拟过程中实时计算质子化状态。在这里,我们报告了基于 GBNeck2 广义 Born(GB)隐溶剂模型在 Amber 分子动力学包的 pmemd 引擎中实现 CpHMD 方法的 CPU 基实现。该方法的性能使用 pH 值 replica-exchange 滴定模拟进行了测试,涉及 4 个小蛋白和 7 种具有实验已知 pK 值的酶中的 Asp、Glu 和 His 侧链,其中一些明显偏离模型值。由于 CpHMD 滴定而增加的计算成本对于数据集在 11%到 33%之间,并且大致与可滴定位点与溶质原子数的比例呈线性关系。使用 2 ns 每个副本采样对实验和计算的 pK 值进行比较,得到无符号误差平均值为 0.70,均方根误差为 0.91,线性相关系数为 0.79。尽管这种精度水平与 CHARMM 中的 GBSW 基 CpHMD 相似,但与后者不同的是,当前实现能够重现实验中耦合羧酸二联体的 pK 值顺序。我们量化了采样误差,这表明需要进行长时间的模拟才能收敛涉及盐桥样相互作用或深埋在蛋白质内部的几个可滴定基团的 pK 值。我们的基准数据表明,GBNeck2-CpHMD 是一种用于蛋白质 pK 值预测的有吸引力的工具。