MacKay Kimberly, Kusalik Anthony, Eskiw Christopher H
Department of Computer Science, University of Saskatchewan, 110 Science Place, Saskatoon, SK, S7N 5C9, Canada.
Department of Food and Bioproduct Science, University of Saskatchewan, 51 Campus Drive, Saskatoon, SK, S7N 5A8, Canada.
BMC Res Notes. 2018 Jun 29;11(1):418. doi: 10.1186/s13104-018-3507-2.
Hi-C is a proximity-based ligation reaction used to detect regions of the genome that are close in 3D space (or "interacting"). Typically, results from Hi-C experiments (contact maps) are visualized as heatmaps or Circos plots. While informative, these visualizations do not directly represent genomic structure and folding, making the interpretation of the underlying 3D genomic organization obscured. Our objective was to generate a graph-based contact map representation that leads to a more intuitive structural visualization.
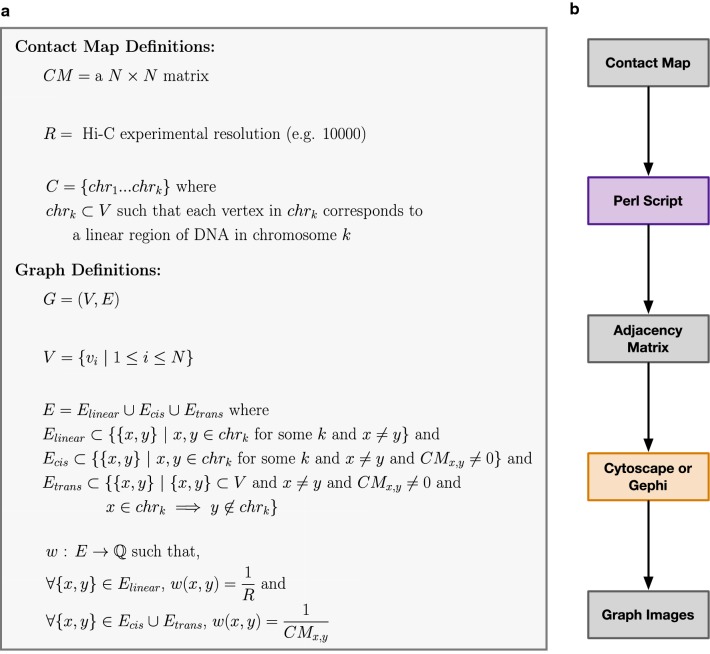
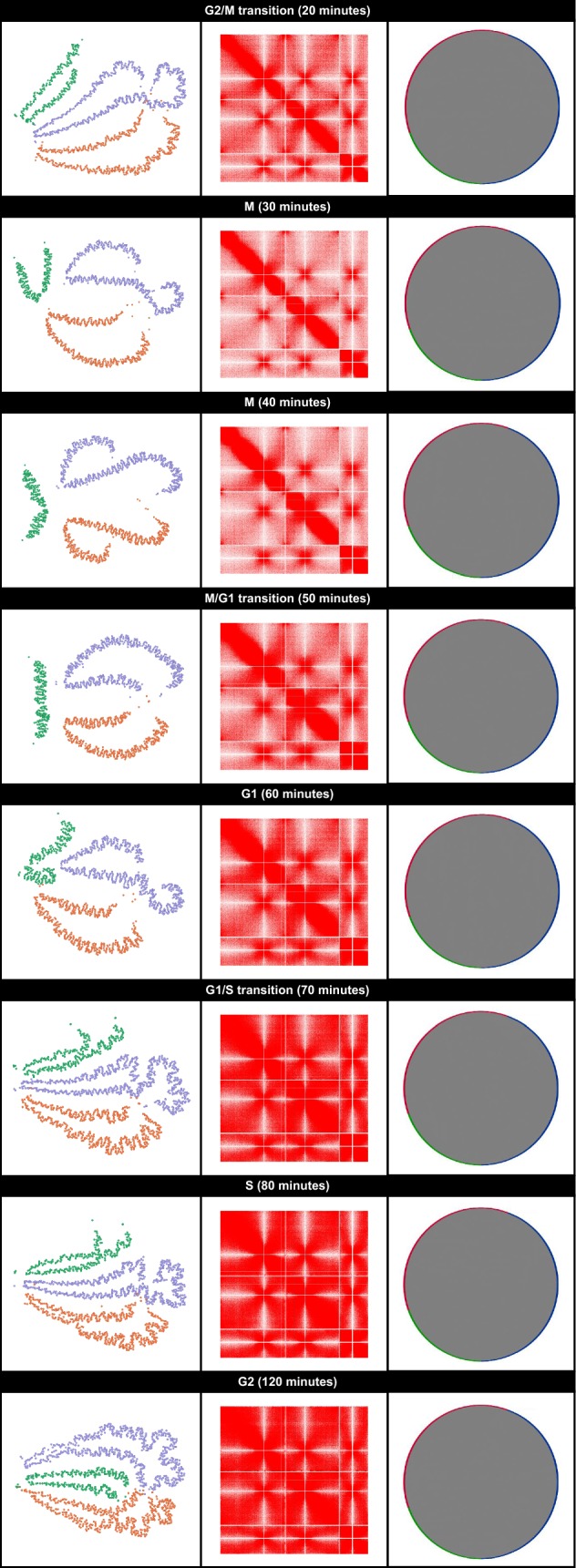
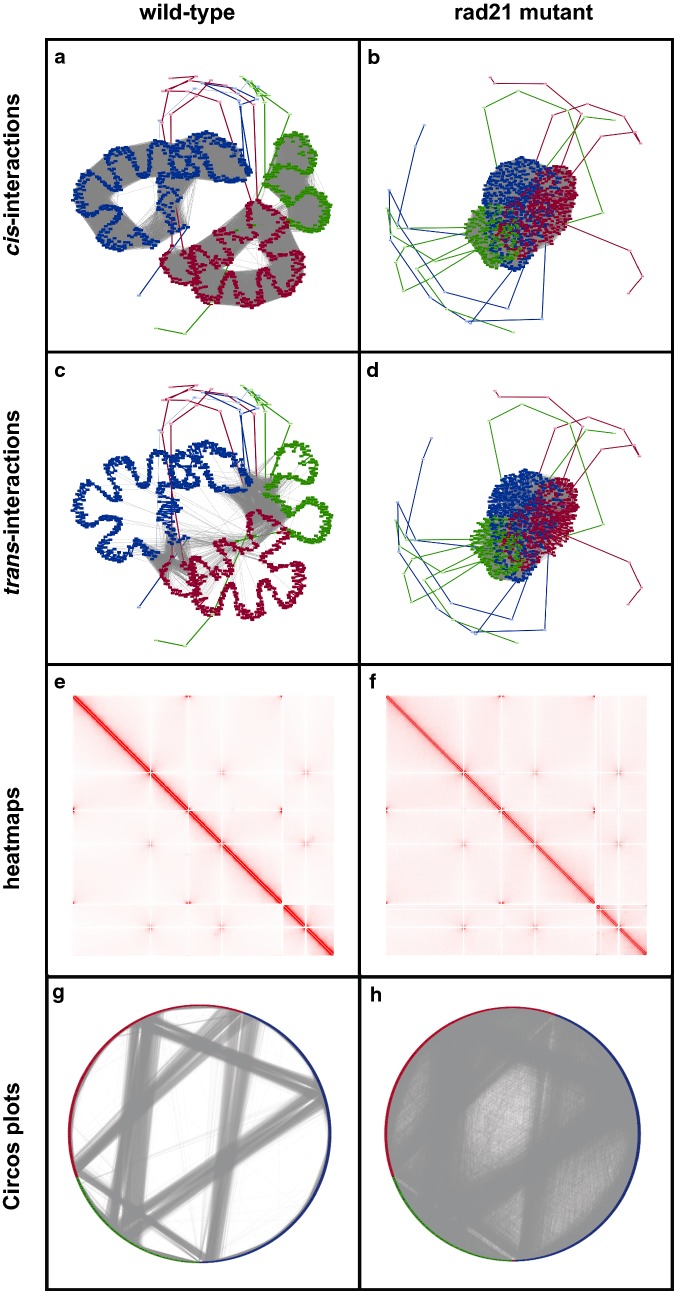
Normalized contact maps were converted into undirected graphs where each vertex represented a genomic region and each edge represented a detected (intra- and inter-chromosomal) or known (linear) interaction between two regions. Each edge was weighted by the inverse of the linear distance (Hi-C experimental resolution) or the interaction frequency from the contact map. Graphs were generated based on this representation scheme for contact maps from existing fission yeast datasets. Originally, these datasets were used to (1) identify specific principles influencing fission yeast genome organization and (2) uncover changes in fission yeast genome organization during the cell cycle. When compared to the equivalent heatmaps and/or Circos plots, the graph-based visualizations more intuitively depicted the changes in genome organization described in the original studies.
Hi-C是一种基于邻近连接反应的技术,用于检测基因组中在三维空间中距离相近(或“相互作用”)的区域。通常,Hi-C实验的结果(接触图谱)以热图或Circos图的形式呈现。虽然这些可视化方式提供了信息,但它们并不能直接代表基因组结构和折叠情况,使得对潜在三维基因组组织的解读变得模糊。我们的目标是生成一种基于图形的接触图谱表示形式,从而实现更直观的结构可视化。
将标准化的接触图谱转换为无向图,其中每个顶点代表一个基因组区域,每条边代表两个区域之间检测到的(染色体内和染色体间)或已知的(线性)相互作用。每条边的权重为线性距离的倒数(Hi-C实验分辨率)或来自接触图谱的相互作用频率。基于这种表示方案,为来自现有裂殖酵母数据集的接触图谱生成了图形。最初,这些数据集用于(1)确定影响裂殖酵母基因组组织的特定原则,以及(2)揭示细胞周期中裂殖酵母基因组组织的变化。与等效的热图和/或Circos图相比,基于图形的可视化更直观地描绘了原始研究中描述的基因组组织变化。