Franco Rafael, Sánchez-Arias Juan A, Navarro Gemma, Lanciego José L
Department of Biochemistry and Molecular Biomedicine, School of Biology, University of Barcelona, Barcelona, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CiberNed), Instituto de Salud Carlos III, Madrid, Spain.
Front Neuroanat. 2018 Jun 28;12:52. doi: 10.3389/fnana.2018.00052. eCollection 2018.
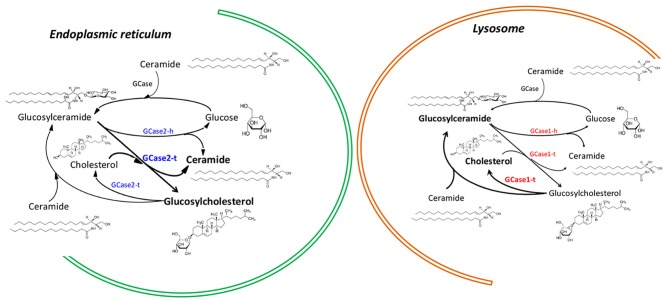
Gaucher's disease (GD) is the most prevalent lysosomal storage disorder. GD is caused by homozygous mutations of the GBA1 gene, which codes for beta-glucocerebrosidase (GCase). Although GD primarily affects peripheral tissues, the presence of neurological symptoms has been reported in several GD subtypes. GBA1 mutations have recently deserved increased attention upon the demonstration that both homo- and heterozygous GBA1 mutations represent the most important genetic risk factor for the appearance of synucleinopathies like Parkinson's disease (PD) and dementia with Lewy bodies (LBD). Although reduced GCase activity leads to alpha-synuclein aggregation, the mechanisms sustaining a role for GCase in alpha-synuclein homeostasis still remain largely unknown. Furthermore, the role to be played by impairment in the physiological function of endoplasmic reticulum, mitochondria and other subcellular membranous components is currently under investigation. Here we focus on the impact of GCase loss-of-function that impact on the levels of sterylglucosides, molecules that are known to trigger a PD-related synucleinopathy upon administration in rats. Moreover, the concurrence of another gene also coding for an enzyme with GCase activity (GBA2 gene) should also be taken into consideration, bearing in mind that in addition to a hydrolytic function, both GCases also share transglycosylation as a second catalytic activity. Accordingly, sterylglycoside levels should also be considered to further assess their impact on the neurodegenerative process. In this regard-and besides GBA1 genotyping-we suggest that screening for GBA2 mutations should be considered, together with analytical measurements of cholesterol glycosides in body fluids, as biomarkers for both PD risk and disease progression.
戈谢病(GD)是最常见的溶酶体贮积症。GD由GBA1基因突变纯合子引起,该基因编码β-葡萄糖脑苷脂酶(GCase)。尽管GD主要影响外周组织,但在几种GD亚型中也有神经症状的报道。最近,GBA1突变受到了更多关注,因为研究表明,GBA1基因的纯合子和杂合子突变都是帕金森病(PD)和路易体痴呆(LBD)等突触核蛋白病出现的最重要遗传风险因素。尽管GCase活性降低会导致α-突触核蛋白聚集,但GCase在α-突触核蛋白稳态中发挥作用的机制仍 largely未知。此外,内质网、线粒体和其他亚细胞膜成分的生理功能受损所起的作用目前正在研究中。在这里,我们关注GCase功能丧失对甾醇糖苷水平的影响,已知在大鼠体内给药时,这些分子会引发与PD相关的突触核蛋白病。此外,还应考虑另一个也编码具有GCase活性的酶的基因(GBA2基因)的存在,要记住,除了水解功能外,两种GCase还都具有转糖基化作为第二种催化活性。因此,甾醇糖苷水平也应被考虑,以进一步评估它们对神经退行性过程的影响。在这方面,除了GBA1基因分型外,我们建议应考虑筛查GBA2突变,并同时对体液中的胆固醇糖苷进行分析测量,将其作为PD风险和疾病进展的生物标志物。