Division of General Internal Medicine and Primary Care, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts, 02120, USA.
Department of Biomedical Informatics, Harvard Medical School, Boston, Massachusetts, 02115, USA.
Sci Rep. 2018 Jul 27;8(1):11360. doi: 10.1038/s41598-018-29634-w.
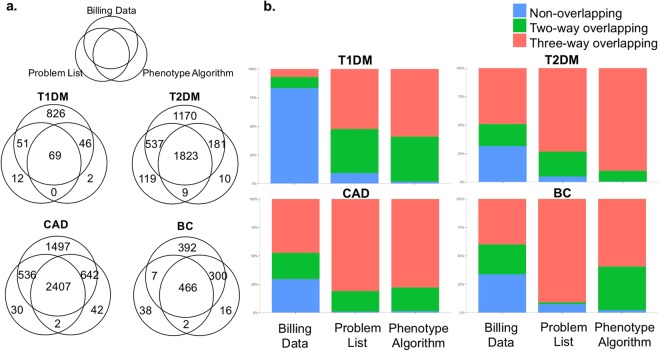
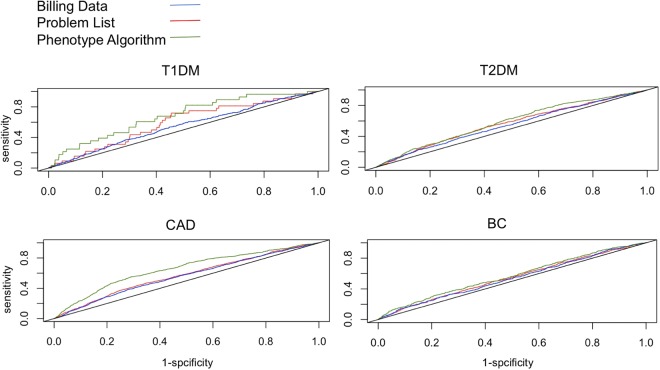
Genome-wide association studies depend on accurate ascertainment of patient phenotype. However, phenotyping is difficult, and it is often treated as an afterthought in these studies because of the expense involved. Electronic health records (EHRs) may provide higher fidelity phenotypes for genomic research than other sources such as administrative data. We used whole genome association models to evaluate different EHR and administrative data-based phenotyping methods in a cohort of 16,858 Caucasian subjects for type 1 diabetes mellitus, type 2 diabetes mellitus, coronary artery disease and breast cancer. For each disease, we trained and evaluated polygenic models using three different phenotype definitions: phenotypes derived from billing data, the clinical problem list, or a curated phenotyping algorithm. We observed that for these diseases, the curated phenotype outperformed the problem list, and the problem list outperformed administrative billing data. This suggests that using advanced EHR-derived phenotypes can further increase the power of genome-wide association studies.
全基因组关联研究依赖于对患者表型的准确确定。然而,表型确定很困难,由于涉及到费用问题,在这些研究中通常被视为事后考虑。电子健康记录 (EHR) 可能为基因组研究提供比其他来源(如行政数据)更高保真度的表型。我们使用全基因组关联模型,在一个包含 16858 名白种人受试者的队列中,对 1 型糖尿病、2 型糖尿病、冠状动脉疾病和乳腺癌的不同 EHR 和基于行政数据的表型方法进行了评估。对于每种疾病,我们使用三种不同的表型定义(从计费数据、临床问题列表或经过精心策划的表型算法中得出的表型)来训练和评估多基因模型。我们观察到,对于这些疾病,经过策划的表型优于问题列表,而问题列表优于行政计费数据。这表明使用先进的基于 EHR 的表型可以进一步提高全基因组关联研究的功效。