Farber-Katz Suzette, Hsuan Vickie, Wu Sitao, Landrith Tyler, Vuong Huy, Xu Dong, Li Bing, Hoo Jayne, Lam Stephanie, Nashed Sarah, Toppmeyer Deborah, Gray Phillip, Haynes Ginger, Lu Hsiao-Mei, Elliott Aaron, Tippin Davis Brigette, Karam Rachid
Translational Genomics Laboratory, Ambry Genetics, Aliso Viejo, CA, United States.
Department of Bioinformatics, Ambry Genetics, Aliso Viejo, CA, United States.
Front Oncol. 2018 Jul 27;8:286. doi: 10.3389/fonc.2018.00286. eCollection 2018.
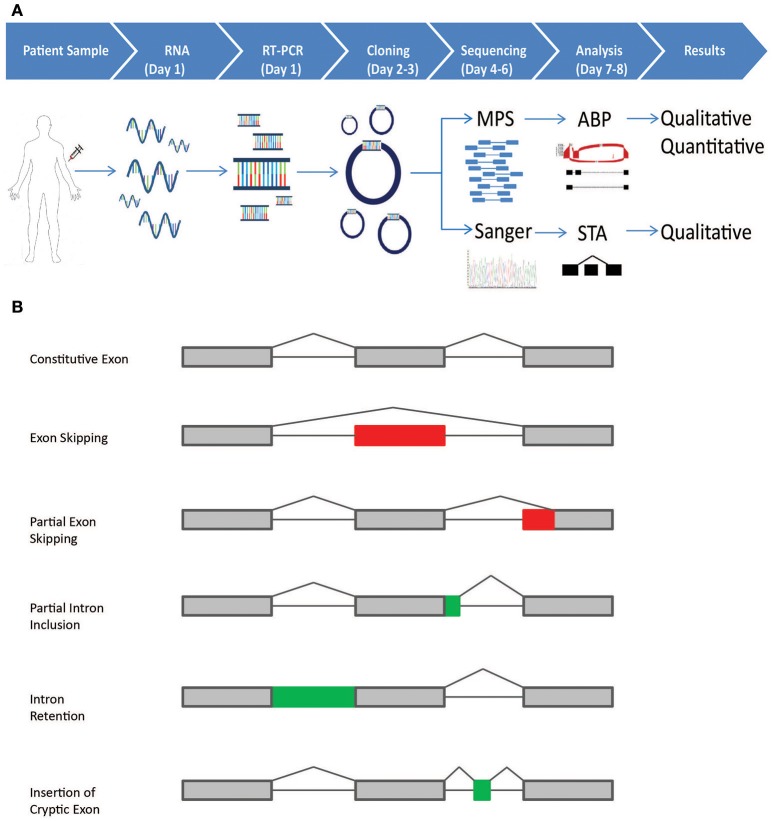
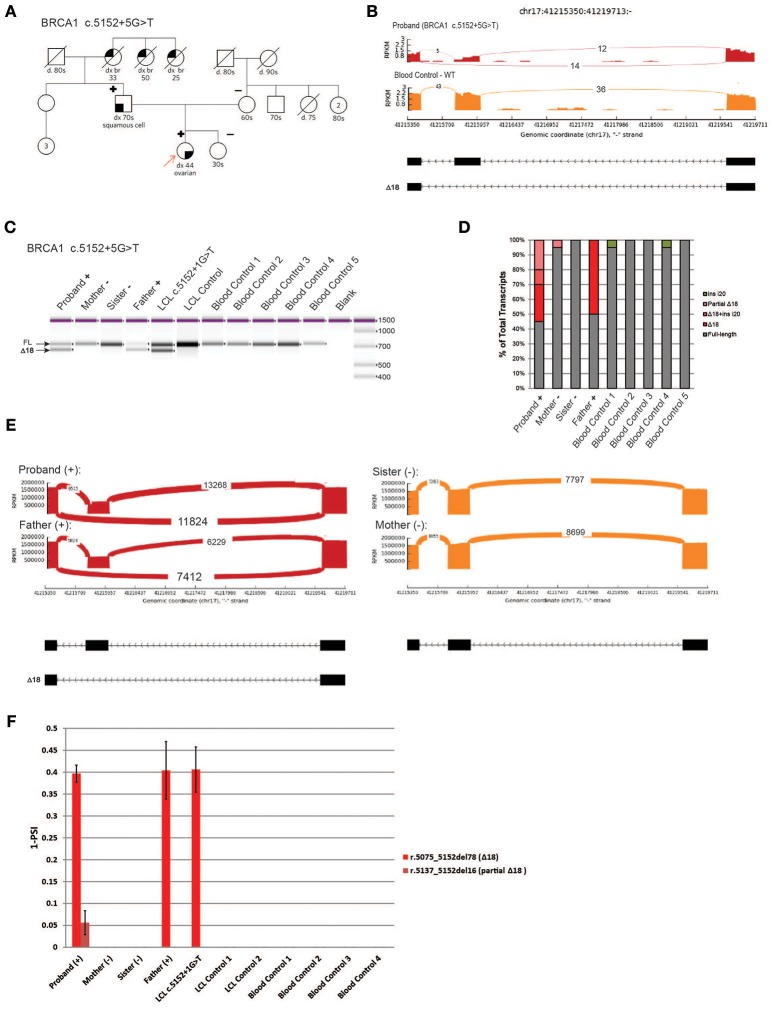
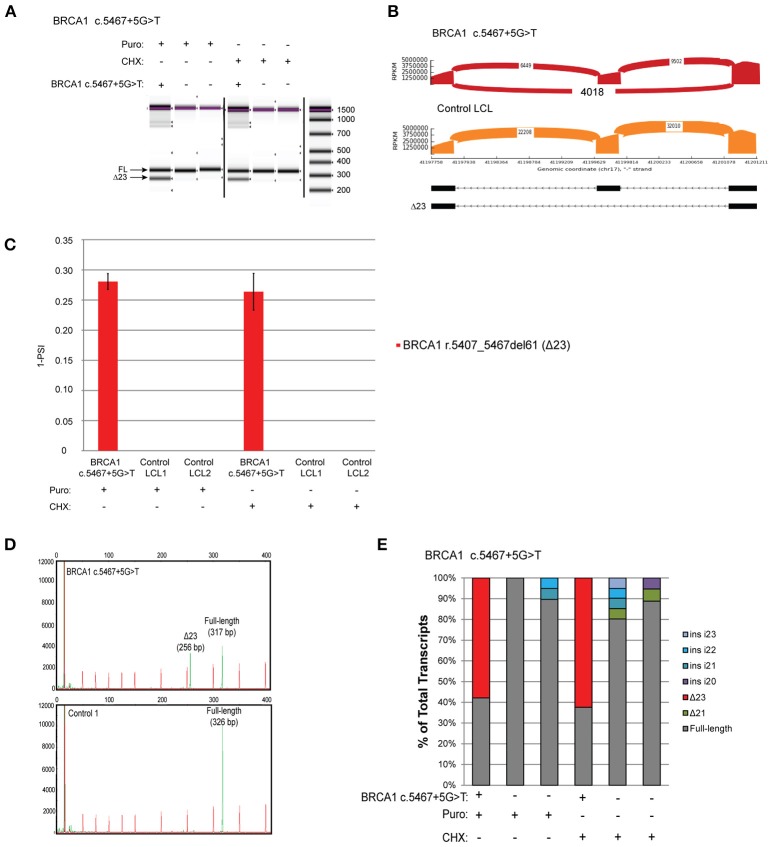
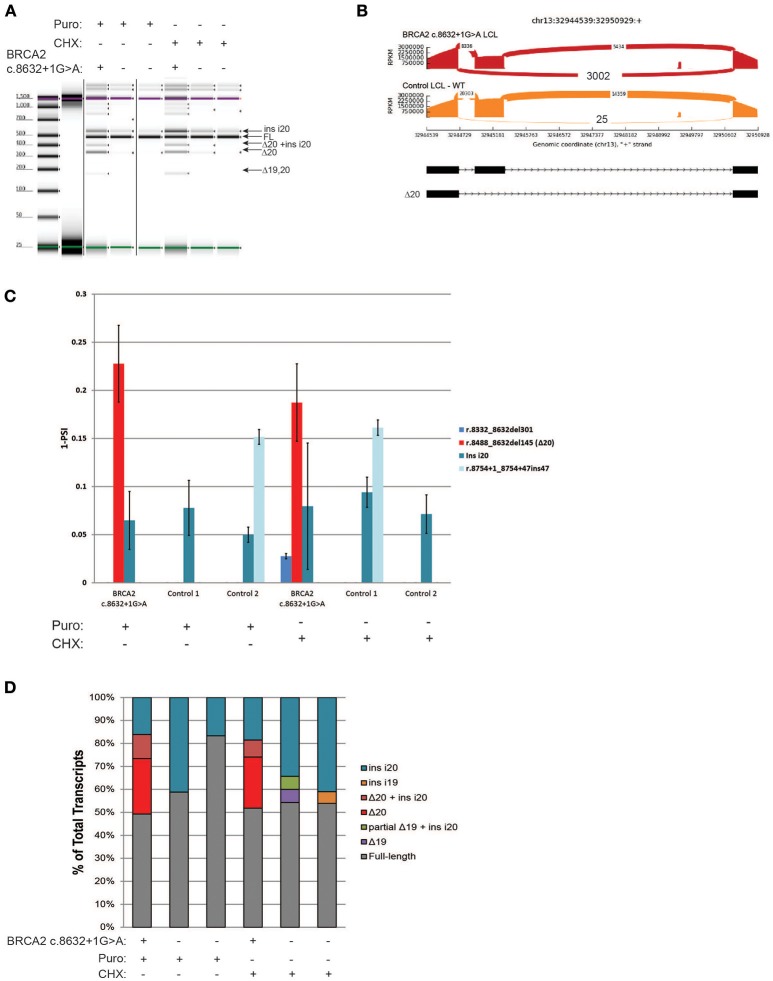
Clinical genetic testing for hereditary breast and ovarian cancer (HBOC) is becoming widespread. However, the interpretation of variants of unknown significance (VUS) in HBOC genes, such as the clinically actionable genes and , remain a challenge. Among the variants that are frequently classified as VUS are those with unclear effects on splicing. In order to address this issue we developed a high-throughput RNA-massively parallel sequencing assay--capable to perform quantitative and qualitative analysis of transcripts in cell lines and HBOC patients. This assay is based on cloning of RT-PCR products followed by massive parallel sequencing of the cloned transcripts. To validate this assay we compared it to the RNA splicing assays recommended by members of the ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) consortium. This comparison was performed using well-characterized lymphoblastoid cell lines (LCLs) generated from carriers of the or germline variants that have been previously described to be associated with splicing defects. CloneSeq was able to replicate the ENIGMA results, in addition to providing quantitative characterization of and germline splicing alterations in a high-throughput fashion. Furthermore, CloneSeq was used to analyze blood samples obtained from carriers of or germline sequence variants, including the novel uncharacterized alteration c.5152+5G>T, which was identified in a HBOC family. CloneSeq provided a high-resolution picture of all the transcripts induced by c.5152+5G>T, indicating it results in significant levels of exon skipping. This analysis proved to be important for the classification of c.5152+5G>T as a clinically actionable likely pathogenic variant. Reclassifications such as these are fundamental in order to offer preventive measures, targeted treatment, and pre-symptomatic screening to the correct individuals.
遗传性乳腺癌和卵巢癌(HBOC)的临床基因检测正日益普及。然而,对HBOC相关基因中意义未明变异(VUS)的解读,如对临床可操作基因 和 的解读,仍然是一项挑战。在经常被归类为VUS的变异中,有一些对剪接的影响尚不清楚。为了解决这个问题,我们开发了一种高通量RNA大规模平行测序检测方法,该方法能够对细胞系和HBOC患者的转录本进行定量和定性分析。此检测方法基于逆转录聚合酶链反应(RT-PCR)产物的克隆,随后对克隆的转录本进行大规模平行测序。为了验证该检测方法,我们将其与由ENIGMA(种系突变等位基因解释循证网络)联盟成员推荐的RNA剪接检测方法进行了比较。这种比较是使用从先前已被描述为与剪接缺陷相关的 或 种系变异携带者产生的特征明确的淋巴母细胞系(LCLs)进行的。除了以高通量方式提供 和 种系剪接改变的定量特征外,CloneSeq还能够重现ENIGMA的结果。此外,CloneSeq被用于分析从 或 种系序列变异携带者获取的血液样本,包括在一个HBOC家族中鉴定出的新型未表征变异c.5152+5G>T。CloneSeq提供了由c.5152+5G>T诱导的所有转录本的高分辨率图谱,表明它导致了显著水平的外显子跳跃。这一分析对于将c.5152+5G>T分类为临床可操作的可能致病变异至关重要。这样的重新分类对于为正确的个体提供预防措施、靶向治疗和症状前筛查至关重要。