Kono Thomas J Y, Lei Li, Shih Ching-Hua, Hoffman Paul J, Morrell Peter L, Fay Justin C
Department of Agronomy & Plant Genetics, University of Minnesota, St. Paul, MN 551085.
Department of Genetics, Washington University, St. Louis, MO 63110.
G3 (Bethesda). 2018 Oct 3;8(10):3321-3329. doi: 10.1534/g3.118.200563.
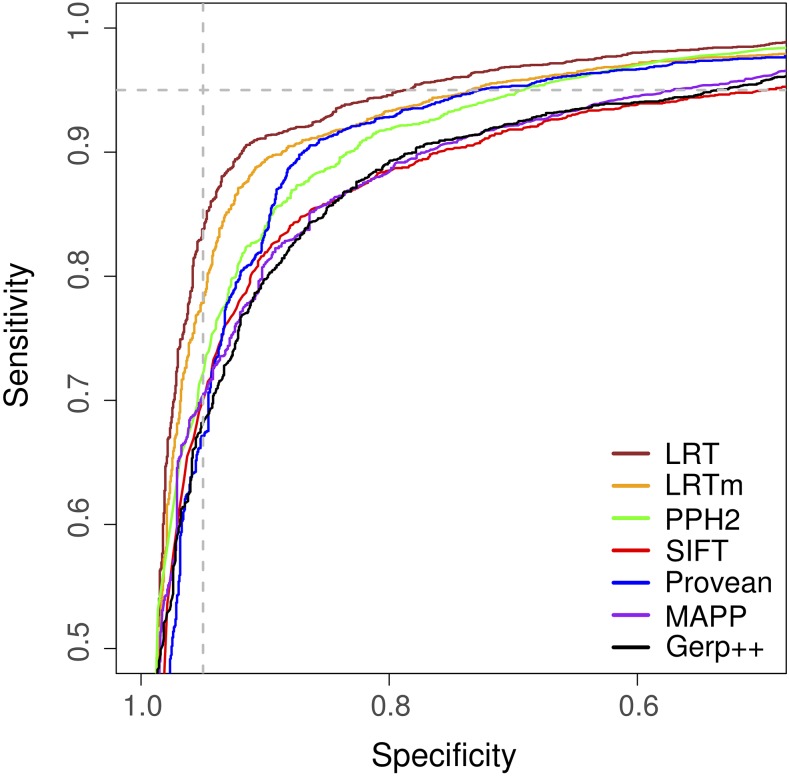
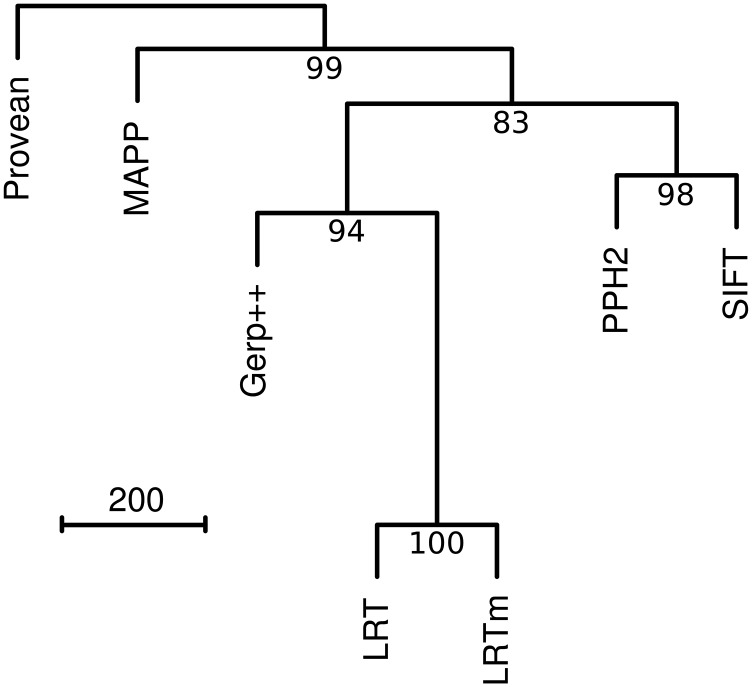
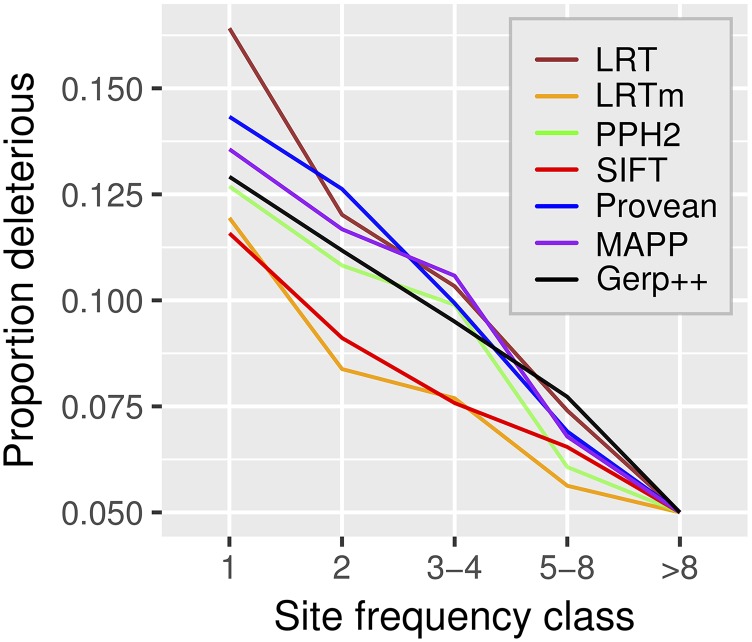
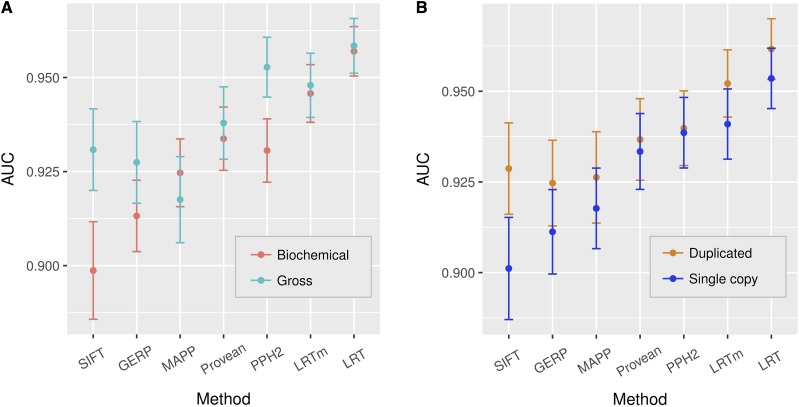
Recent advances in genome resequencing have led to increased interest in prediction of the functional consequences of genetic variants. Variants at phylogenetically conserved sites are of particular interest, because they are more likely than variants at phylogenetically variable sites to have deleterious effects on fitness and contribute to phenotypic variation. Numerous comparative genomic approaches have been developed to predict deleterious variants, but the approaches are nearly always assessed based on their ability to identify known disease-causing mutations in humans. Determining the accuracy of deleterious variant predictions in nonhuman species is important to understanding evolution, domestication, and potentially to improving crop quality and yield. To examine our ability to predict deleterious variants in plants we generated a curated database of 2,910 mutants with known phenotypes. We evaluated seven approaches and found that while all performed well, their relative ranking differed from prior benchmarks in humans. We conclude that deleterious mutations can be reliably predicted in and likely other plant species, but that the relative performance of various approaches does not necessarily translate from one species to another.
基因组重测序技术的最新进展引发了人们对预测基因变异功能后果的更多关注。系统发育保守位点的变异尤其令人感兴趣,因为与系统发育可变位点的变异相比,它们对适应性产生有害影响并导致表型变异的可能性更大。已经开发了许多比较基因组学方法来预测有害变异,但这些方法几乎总是根据其识别人类已知致病突变的能力来评估。确定非人类物种中有害变异预测的准确性对于理解进化、驯化以及潜在地改善作物质量和产量至关重要。为了检验我们预测植物中有害变异的能力,我们生成了一个包含2910个具有已知表型的突变体的精选数据库。我们评估了七种方法,发现虽然所有方法都表现良好,但它们的相对排名与之前人类的基准不同。我们得出结论,有害突变可以在[具体植物物种]以及可能的其他植物物种中可靠地预测,但各种方法的相对性能不一定能从一个物种转移到另一个物种。