Laboratory of Molecular Gerontology, Biomedical Research Center, 251 Bayview Boulevard, National Institute on Aging, NIH, Baltimore, Maryland 21224, USA.
Nat Commun. 2016 Dec 6;7:13785. doi: 10.1038/ncomms13785.
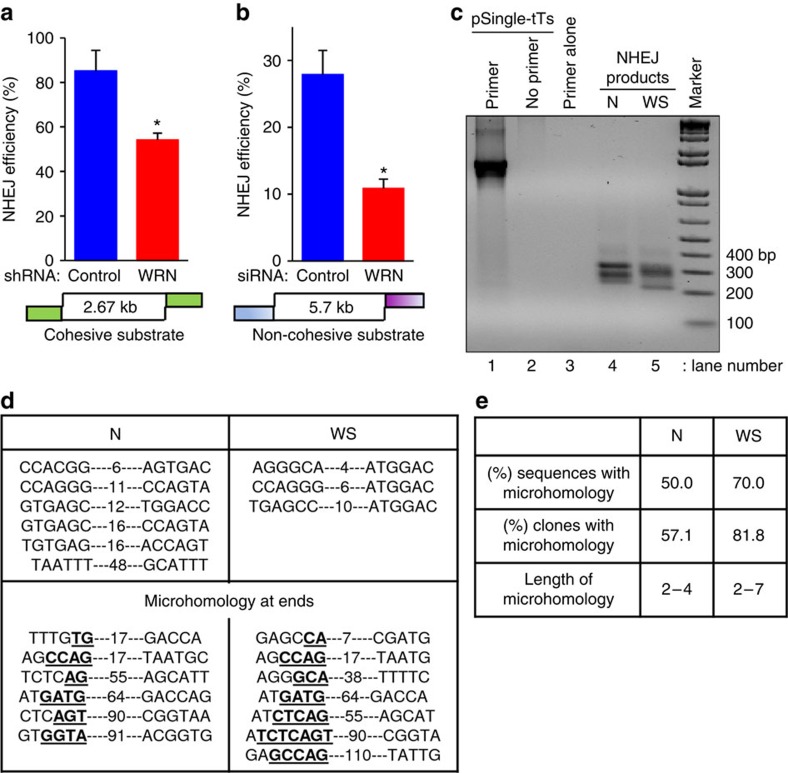
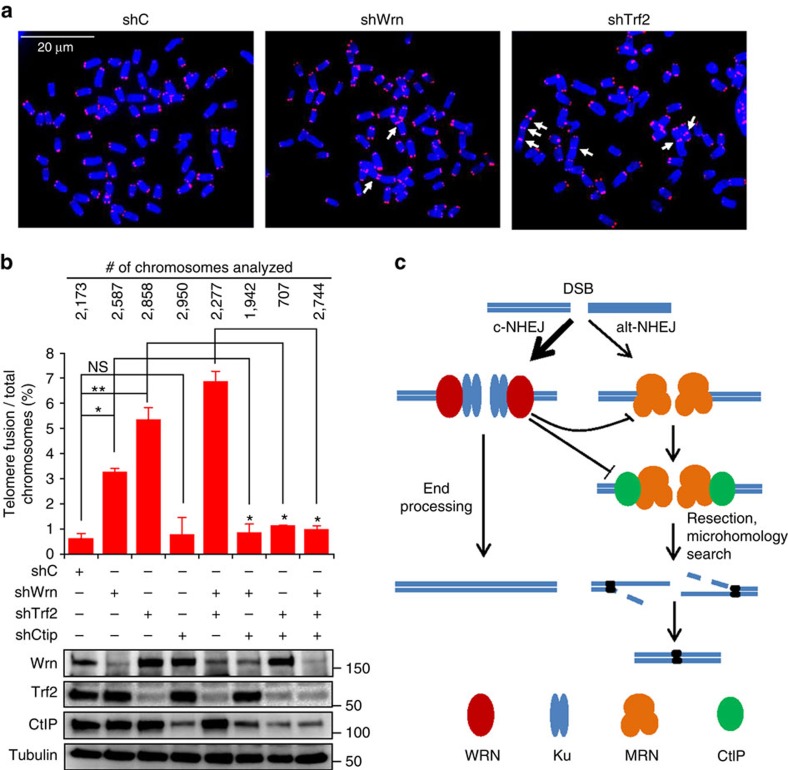
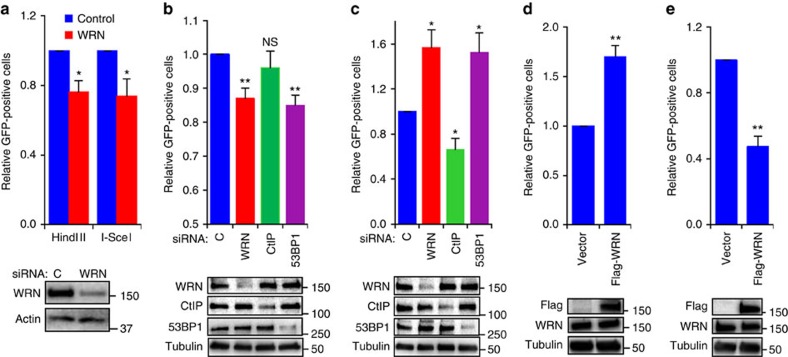
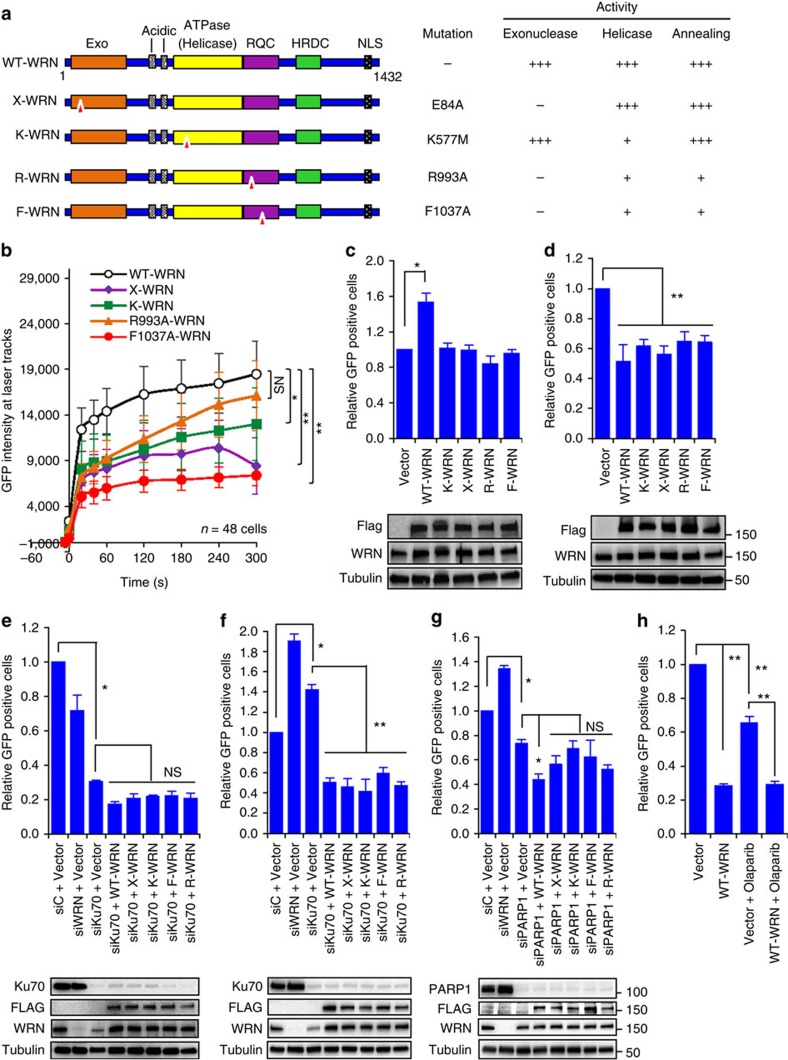
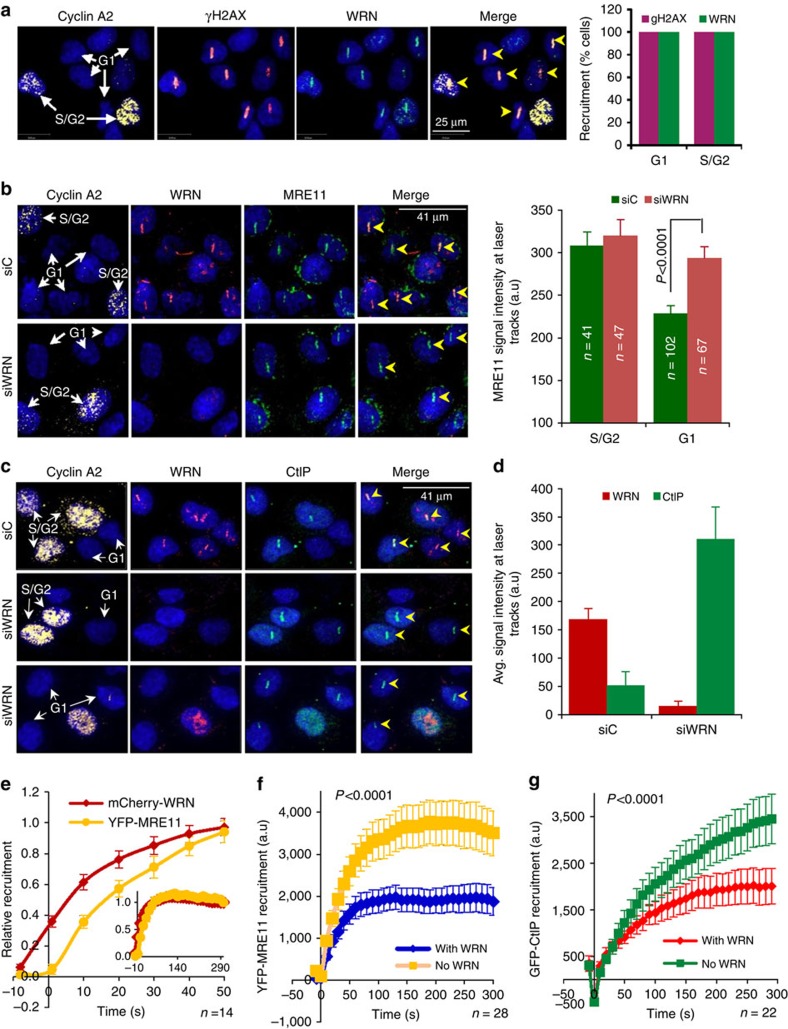
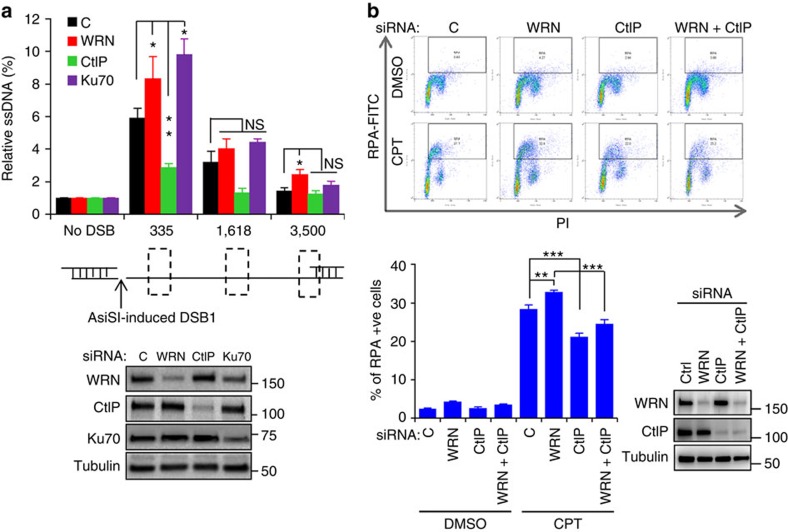
Werner syndrome (WS) is an accelerated ageing disorder with genomic instability caused by WRN protein deficiency. Many features seen in WS can be explained by the diverse functions of WRN in DNA metabolism. However, the origin of the large genomic deletions and telomere fusions are not yet understood. Here, we report that WRN regulates the pathway choice between classical (c)- and alternative (alt)-nonhomologous end joining (NHEJ) during DNA double-strand break (DSB) repair. It promotes c-NHEJ via helicase and exonuclease activities and inhibits alt-NHEJ using non-enzymatic functions. When WRN is recruited to the DSBs it suppresses the recruitment of MRE11 and CtIP, and protects the DSBs from 5' end resection. Moreover, knockdown of Wrn, alone or in combination with Trf2 in mouse embryonic fibroblasts results in increased telomere fusions, which were ablated by Ctip knockdown. We show that WRN regulates alt-NHEJ and shields DSBs from MRE11/CtIP-mediated resection to prevent large deletions and telomere fusions.
Werner 综合征(WS)是一种由 WRN 蛋白缺乏引起的加速老化疾病,具有基因组不稳定性。WS 中观察到的许多特征可以用 WRN 在 DNA 代谢中的多种功能来解释。然而,大的基因组缺失和端粒融合的起源尚不清楚。在这里,我们报告 WRN 调节 DNA 双链断裂(DSB)修复过程中经典(c)-和替代(alt)-非同源末端连接(NHEJ)之间的途径选择。它通过解旋酶和外切核酸酶活性促进 c-NHEJ,并利用非酶功能抑制 alt-NHEJ。当 WRN 被募集到 DSB 时,它会抑制 MRE11 和 CtIP 的募集,并保护 DSB 免受 5' 端切除。此外,在小鼠胚胎成纤维细胞中单独或与 Trf2 联合敲低 Wrn 会导致端粒融合增加,而 Ctip 敲低可消除端粒融合。我们表明,WRN 调节 alt-NHEJ 并保护 DSB 免受 MRE11/CtIP 介导的切除,以防止大的缺失和端粒融合。