Nguyen Phuong Thao, Nguyen Dung, Chea Chanbora, Miyauchi Mutsumi, Fujii Makiko, Takata Takashi
Department of Oral and Maxillofacial Pathobiology, Basic Life Science, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan.
Department of General Internal Medicine, Hiroshima University Hospital, Hiroshima, Japan.
Oncotarget. 2018 Jul 31;9(59):31516-31530. doi: 10.18632/oncotarget.25846.
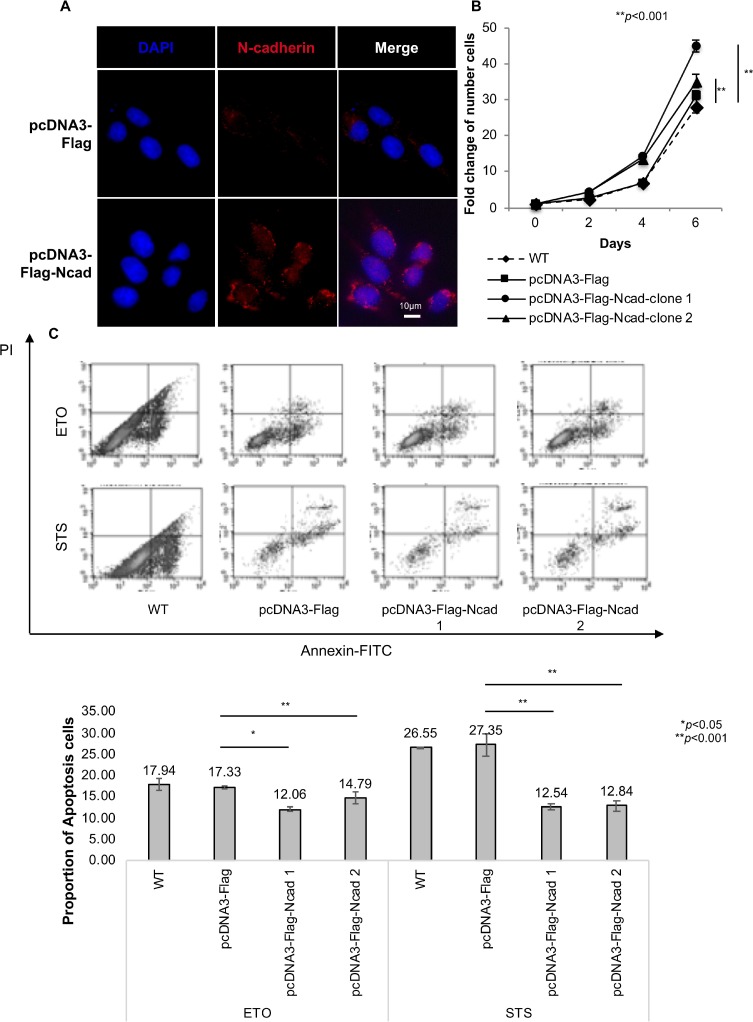
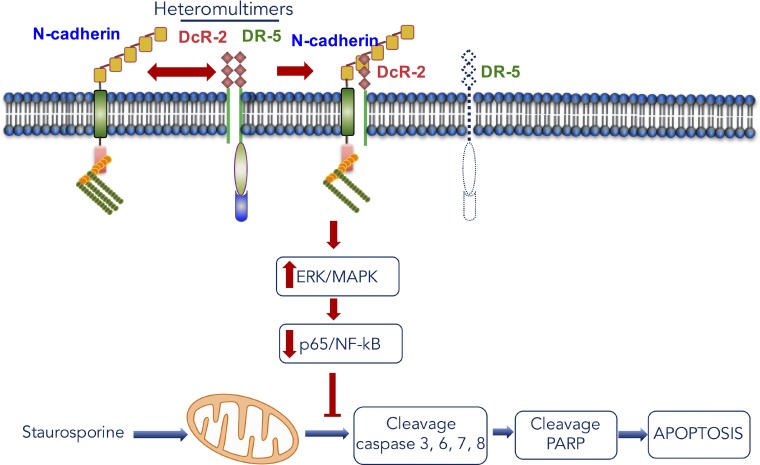
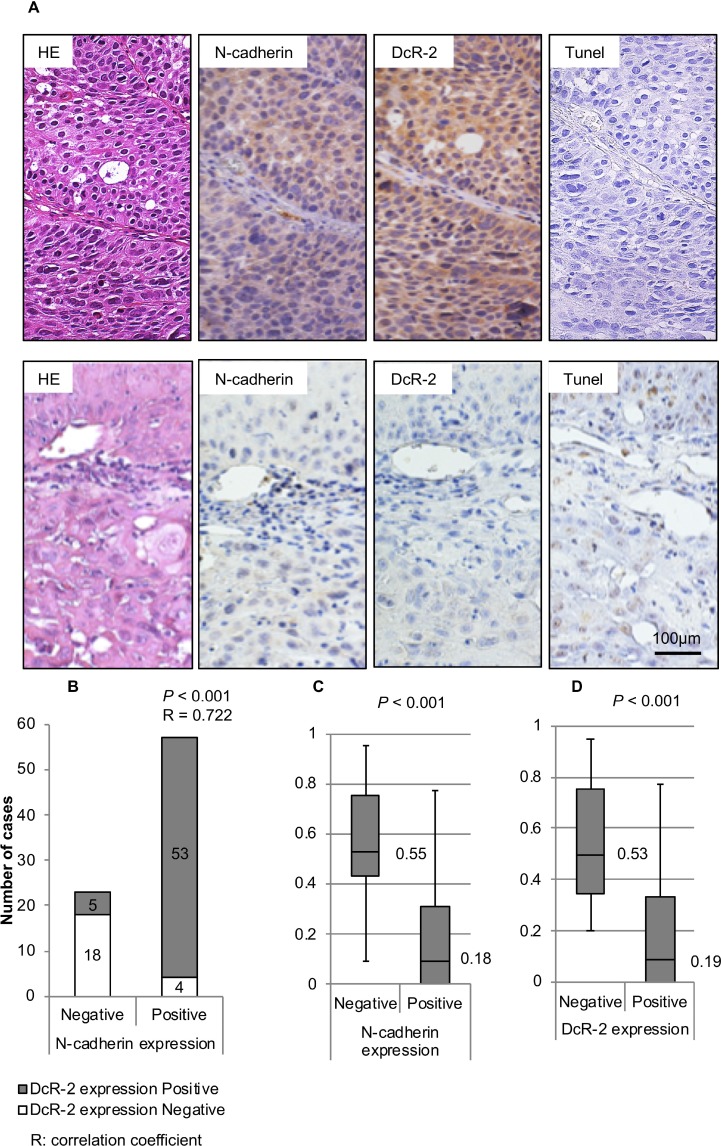
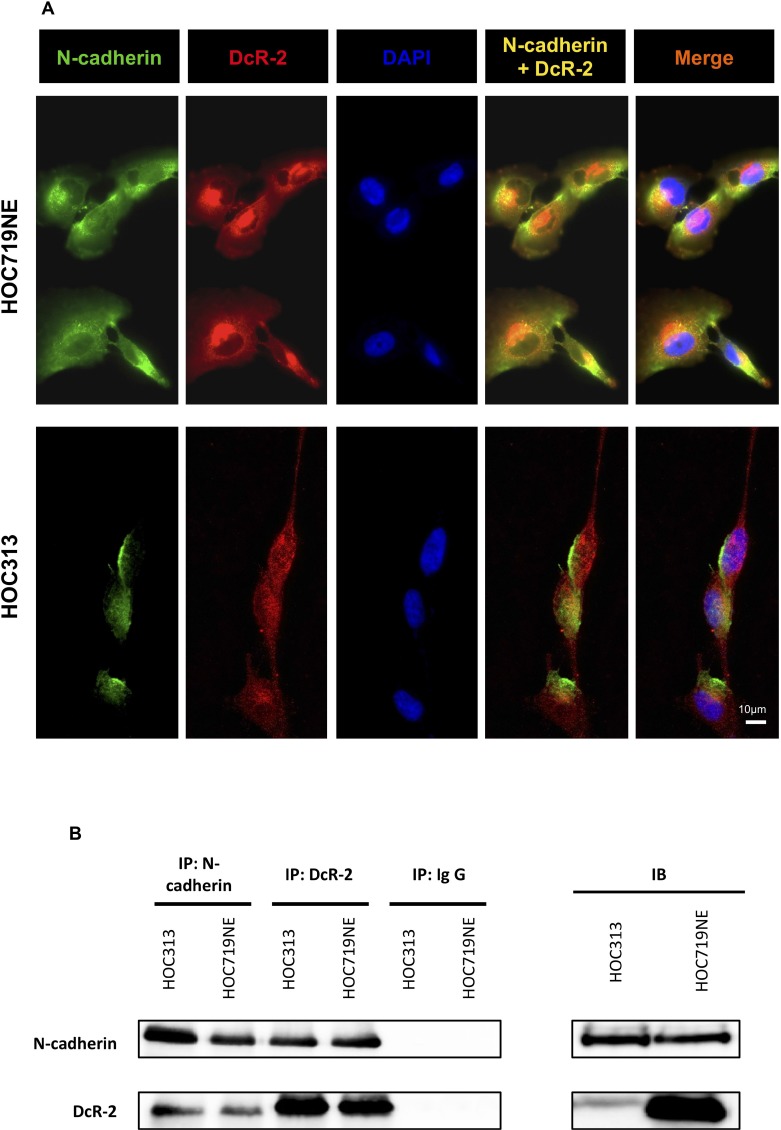
N-cadherin is a neural cell adhesion molecule that aberrantly occurs in head and neck cancers to promote cancer cell growth. However, the underlying mechanisms remain unclear. Here we report that N-cadherin increases cancer cell growth by inhibiting apoptosis. Apoptosis eliminates old, unnecessary, and unhealthy cells. However, tumor cells have the ability of avoiding apoptosis that increases cancer cell growth. Recent studies have found that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) selectively induces apoptosis in tumor cells by reacting with four distinct cell surface receptors: TRAIL-R1 (DR-4), TRAIL-R2 (DR-5), TRAIL-R3 (DcR-1), and TRAIL-R4 (DcR-2). Among these TRAIL receptors, the death receptors DR-4 and DR-5 transmit apoptotic signals owing to the death domain in the intracellular portion. Conversely, the decoy receptors DcR-1 and DcR-2 lack a complete intracellular portion, so neither can transmit apoptotic signals. DcR-1 or DcR-2 overexpression suppresses TRAIL-induced apoptosis. In this study, N-cadherin overexpression increased DcR-2 expression and decreased DR-5 expression. In contrast, knockdown of N-cadherin expression upregulated DR-5 expression and downregulated DcR-2 expression. A significantly positive relationship between N-cadherin and DcR-2 expression was also found in HNSCC specimens. Those specimens with a lower apoptotic index showed a higher expression of N-cadherin and/or DcR-2. In addition, we demonstrated that N-cadherin interacts directly with DcR-2. Notably, DcR-2 induces cancer cell survival through the cleavage of caspases and PARP by activating MAPK/ERK pathway and suppressing NF-kB/ p65 phosphorylation, which has a very important role in resistance to chemotherapy.
N-钙黏蛋白是一种神经细胞黏附分子,在头颈癌中异常表达,可促进癌细胞生长。然而,其潜在机制尚不清楚。在此我们报告,N-钙黏蛋白通过抑制细胞凋亡来促进癌细胞生长。细胞凋亡可清除衰老、不必要和不健康的细胞。然而,肿瘤细胞具有避免细胞凋亡的能力,这会增加癌细胞的生长。最近的研究发现,肿瘤坏死因子相关凋亡诱导配体(TRAIL)通过与四种不同的细胞表面受体反应,选择性地诱导肿瘤细胞凋亡:TRAIL-R1(DR-4)、TRAIL-R2(DR-5)、TRAIL-R3(DcR-1)和TRAIL-R4(DcR-2)。在这些TRAIL受体中,死亡受体DR-4和DR-5由于细胞内部分的死亡结构域而传递凋亡信号。相反,诱饵受体DcR-1和DcR-2缺乏完整的细胞内部分,因此两者都不能传递凋亡信号。DcR-1或DcR-2的过表达会抑制TRAIL诱导的细胞凋亡。在本研究中,N-钙黏蛋白的过表达增加了DcR-2的表达,降低了DR-5的表达。相反,敲低N-钙黏蛋白的表达会上调DR-5的表达,下调DcR-2的表达。在头颈部鳞状细胞癌(HNSCC)标本中也发现N-钙黏蛋白与DcR-2表达之间存在显著的正相关。那些凋亡指数较低的标本显示出较高的N-钙黏蛋白和/或DcR-2表达。此外,我们证明N-钙黏蛋白直接与DcR-2相互作用。值得注意的是,DcR-2通过激活MAPK/ERK途径并抑制NF-κB/p65磷酸化来诱导癌细胞存活,这在化疗耐药中起着非常重要的作用,该过程通过半胱天冬酶和PARP的裂解来实现。