Center for Neuroscience and Regenerative Medicine.
Program in Neuroscience, F. Edward Hebert School of Medicine, Uniformed Services University of the Health Sciences, Bethesda, Maryland 20814.
J Neurosci. 2018 Oct 10;38(41):8723-8736. doi: 10.1523/JNEUROSCI.0819-18.2018. Epub 2018 Aug 24.
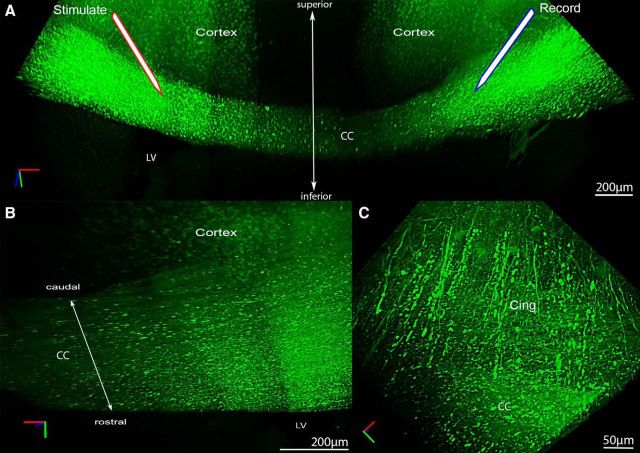
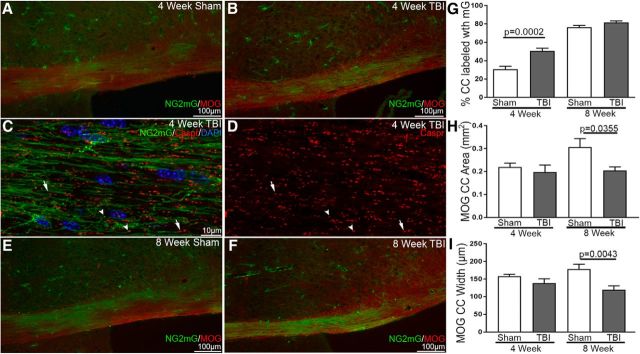
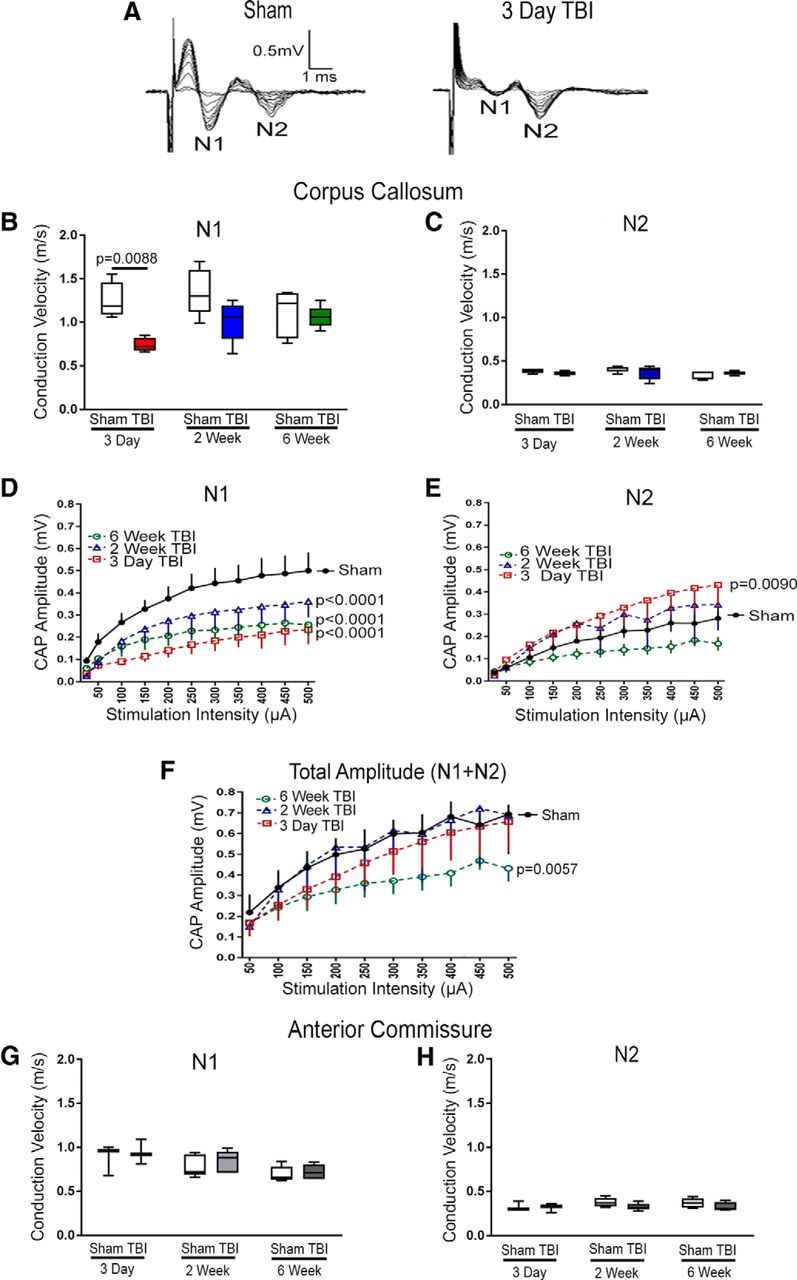
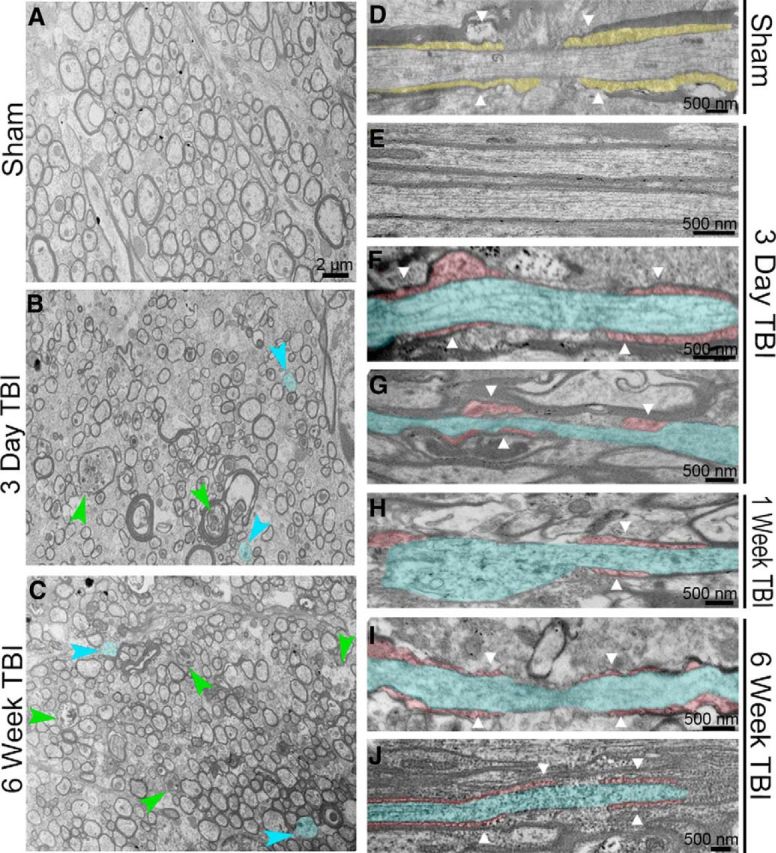
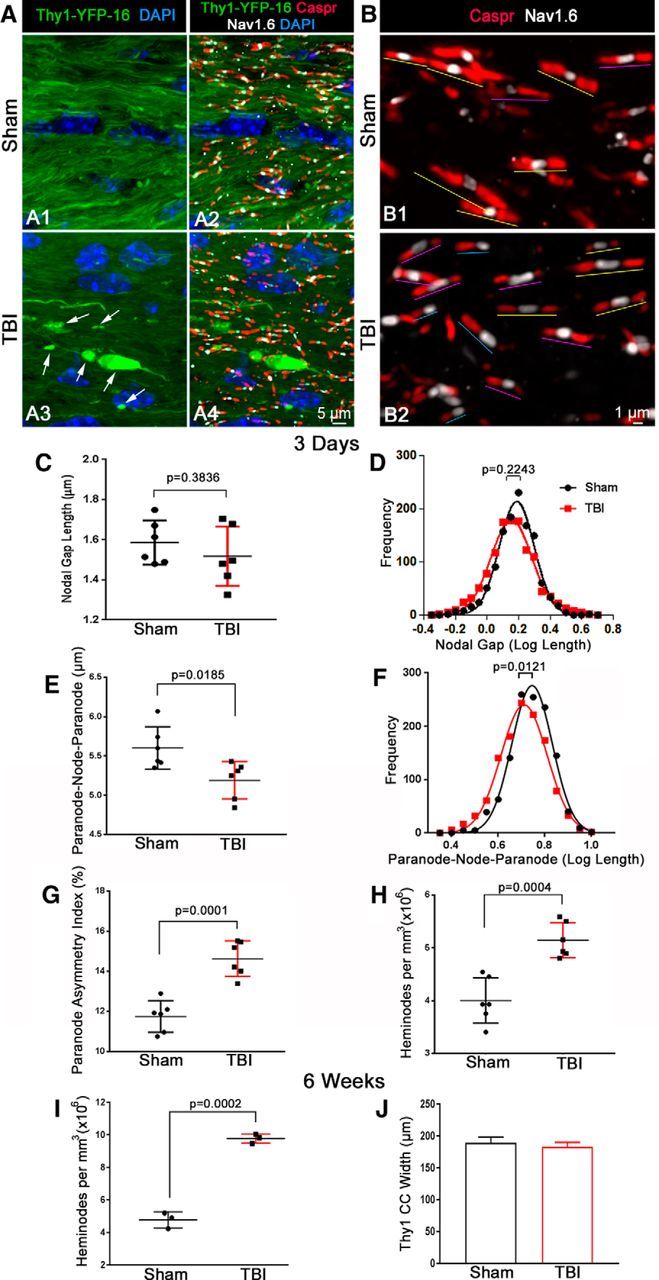
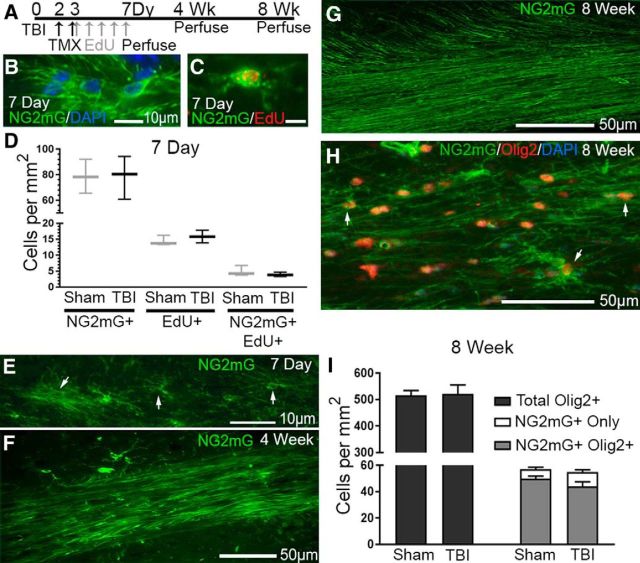
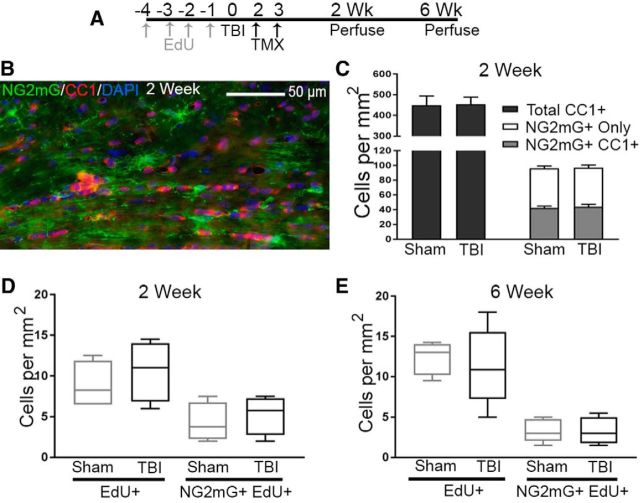
Traumatic brain injury (TBI) patients often exhibit slowed information processing speed that can underlie diverse symptoms. Processing speed depends on neural circuit function at synapses, in the soma, and along axons. Long axons in white matter (WM) tracts are particularly vulnerable to TBI. We hypothesized that disrupted axon-myelin interactions that slow or block action potential conduction in WM tracts may contribute to slowed processing speed after TBI. Concussive TBI in male/female mice was used to produce traumatic axonal injury in the corpus callosum (CC), similar to WM pathology in human TBI cases. Compound action potential velocity was slowed along myelinated axons at 3 d after TBI with partial recovery by 2 weeks, suggesting early demyelination followed by remyelination. Ultrastructurally, dispersed demyelinated axons and disorganized myelin attachment to axons at paranodes were apparent within CC regions exhibiting traumatic axonal injury. Action potential conduction is exquisitely sensitive to paranode abnormalities. Molecular identification of paranodes and nodes of Ranvier detected asymmetrical paranode pairs and abnormal heminodes after TBI. Fluorescent labeling of oligodendrocyte progenitors in mice showed increased synthesis of new membranes extended along axons to paranodes, indicating remyelination after TBI. At later times after TBI, an overall loss of conducting axons was observed at 6 weeks followed by CC atrophy at 8 weeks. These studies identify a progression of both myelinated axon conduction deficits and axon-myelin pathology in the CC, implicating WM injury in impaired information processing at early and late phases after TBI. Furthermore, the intervening recovery reveals a potential therapeutic window. Traumatic brain injury (TBI) is a major global health concern. Across the spectrum of TBI severities, impaired information processing can contribute to diverse functional deficits that underlie persistent symptoms. We used experimental TBI to exploit technical advantages in mice while modeling traumatic axonal injury in white matter tracts, which is a key pathological feature of human TBI. A combination of approaches revealed slowed and failed signal conduction along with damage to the structure and molecular composition of myelinated axons in the white matter after TBI. An early regenerative response was not sustained yet reveals a potential time window for intervention. These insights into white matter abnormalities underlying axon conduction deficits can inform strategies to improve treatment options for TBI patients.
创伤性脑损伤(TBI)患者常表现出信息处理速度减慢,这可能是多种症状的基础。处理速度取决于突触、胞体和轴突中的神经回路功能。白质(WM)束中的长轴突特别容易受到 TBI 的影响。我们假设,破坏轴突-髓鞘相互作用,从而减缓或阻断 WM 束中的动作电位传导,可能是 TBI 后处理速度减慢的原因。雄性/雌性小鼠的震荡性 TBI 用于在胼胝体(CC)中产生创伤性轴突损伤,类似于人类 TBI 病例中的 WM 病理学。TBI 后 3 天,复合动作电位速度在髓鞘化轴突上减慢,2 周时部分恢复,表明早期脱髓鞘后再髓鞘化。超微结构显示,在胼胝体区域显示创伤性轴突损伤的部位,分散的脱髓鞘轴突和轴突旁节排列紊乱的髓鞘附着。动作电位传导对轴突旁节异常非常敏感。TBI 后,在节点和轴突旁节的分子鉴定中检测到不对称的轴突旁节对和异常的半节点。在 小鼠中,少突胶质前体细胞的荧光标记显示,新的膜沿着轴突延伸到轴突旁节,表明 TBI 后有髓鞘形成。TBI 后较晚时间,6 周时观察到传导轴突的总体丢失,8 周时出现胼胝体萎缩。这些研究在胼胝体中确定了髓鞘化轴突传导缺陷和轴突-髓鞘病理的进展,表明 WM 损伤在 TBI 后早期和晚期信息处理受损中起作用。此外,介入恢复揭示了潜在的治疗窗口。创伤性脑损伤(TBI)是一个全球性的主要健康问题。在 TBI 严重程度的范围内,信息处理受损可能导致多种功能缺陷,这些缺陷是持续症状的基础。我们使用实验性 TBI 来利用小鼠的技术优势,同时模拟白质束中的创伤性轴突损伤,这是人类 TBI 的一个关键病理特征。综合方法揭示了 TBI 后白质中信号传导减慢和失败,以及髓鞘化轴突的结构和分子组成受损。早期的再生反应没有持续下去,但揭示了一个潜在的干预时间窗口。这些对 WM 异常导致轴突传导缺陷的见解,可以为改善 TBI 患者的治疗选择提供信息。