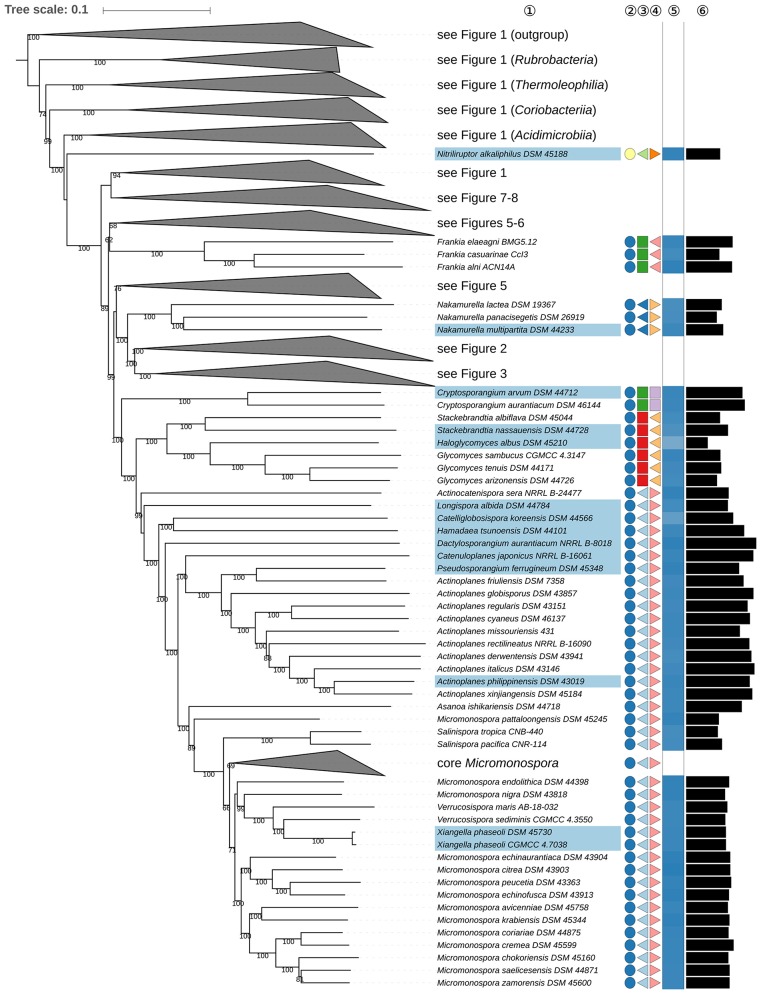
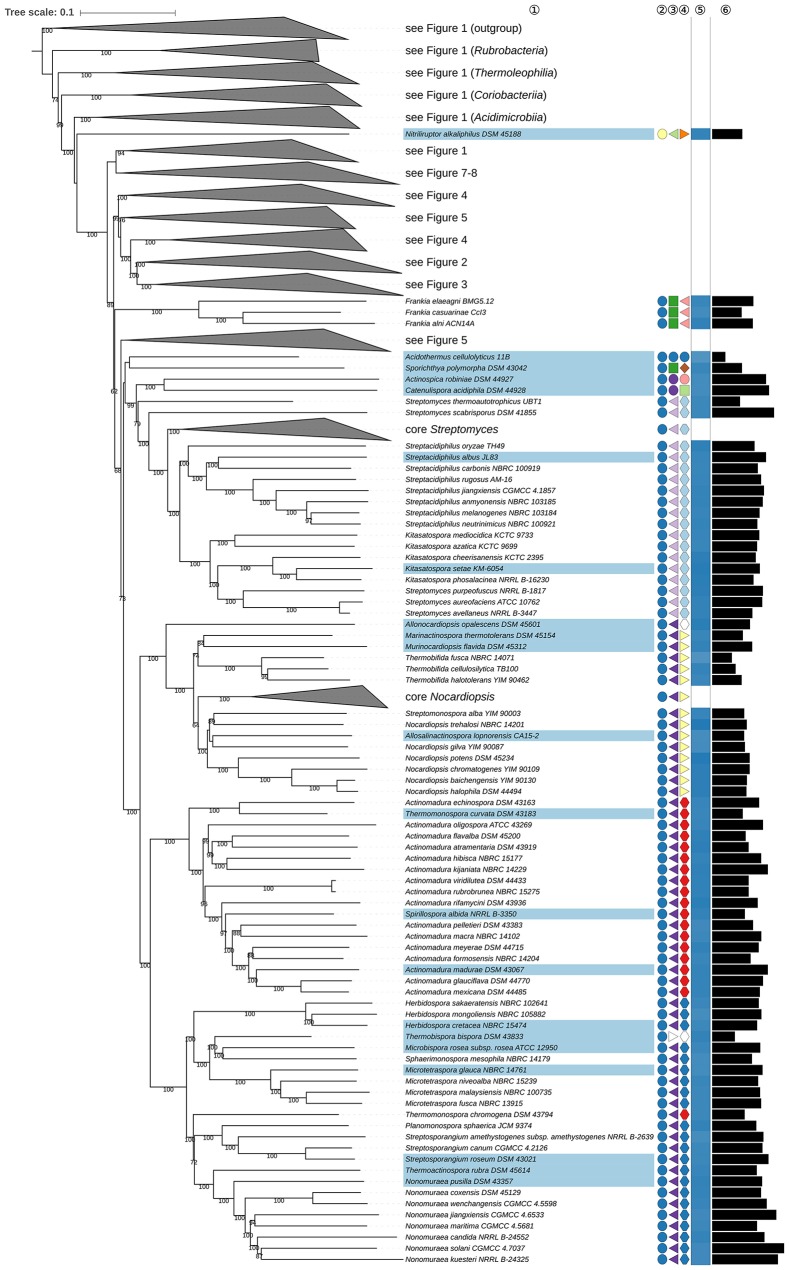
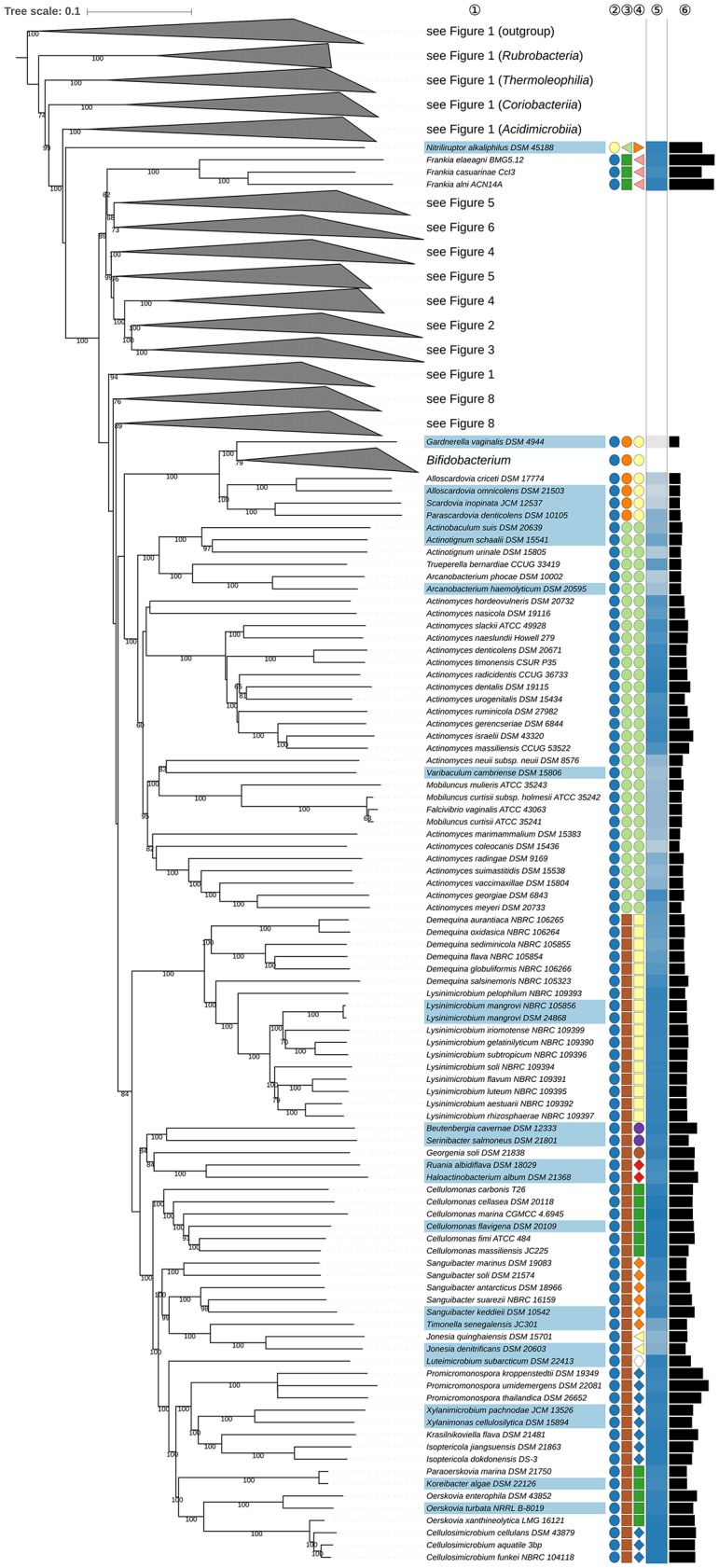
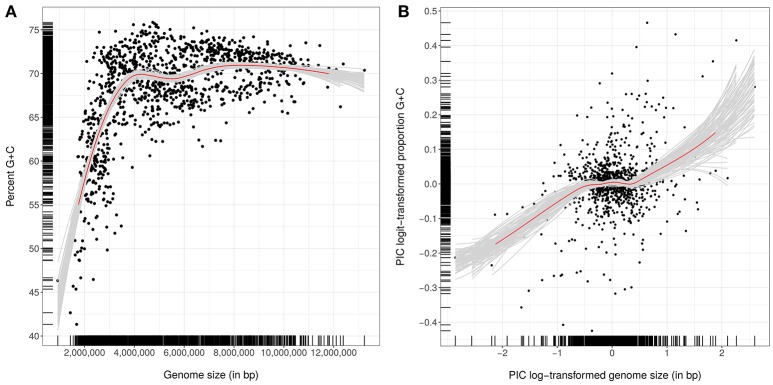
Nouioui Imen, Carro Lorena, García-López Marina, Meier-Kolthoff Jan P, Woyke Tanja, Kyrpides Nikos C, Pukall Rüdiger, Klenk Hans-Peter, Goodfellow Michael, Göker Markus
School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom.
Department of Microorganisms, Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany.
Front Microbiol. 2018 Aug 22;9:2007. doi: 10.3389/fmicb.2018.02007. eCollection 2018.
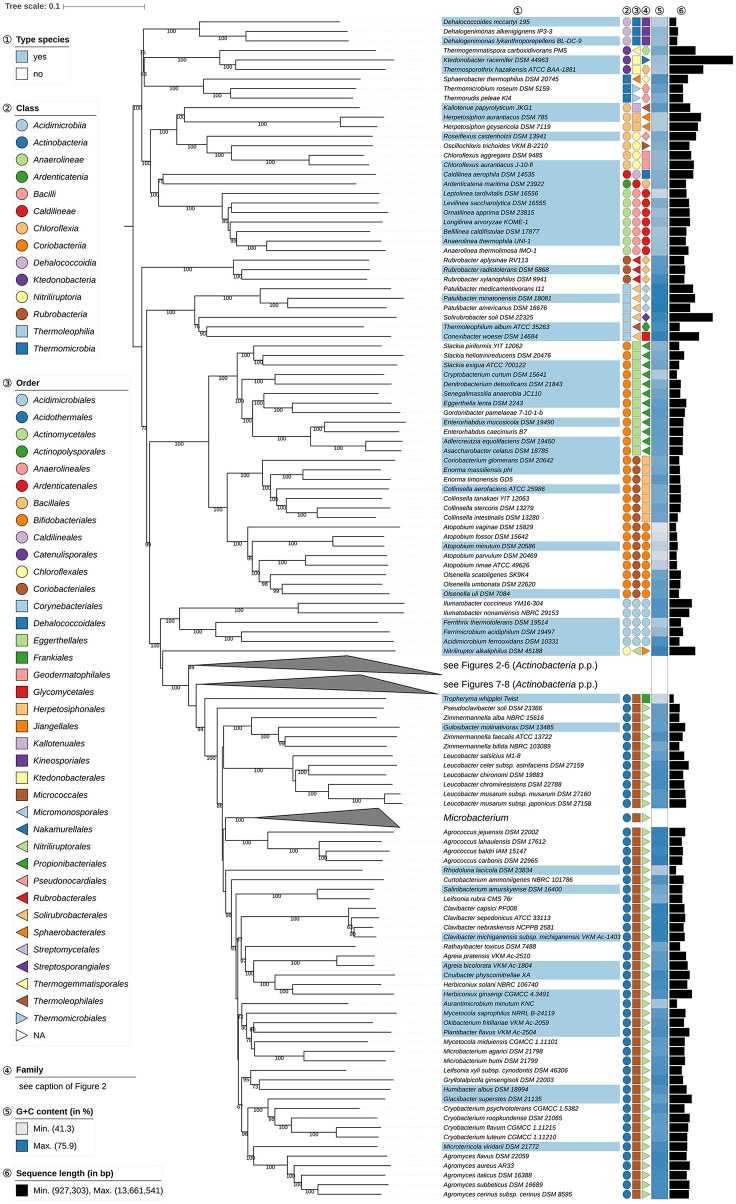
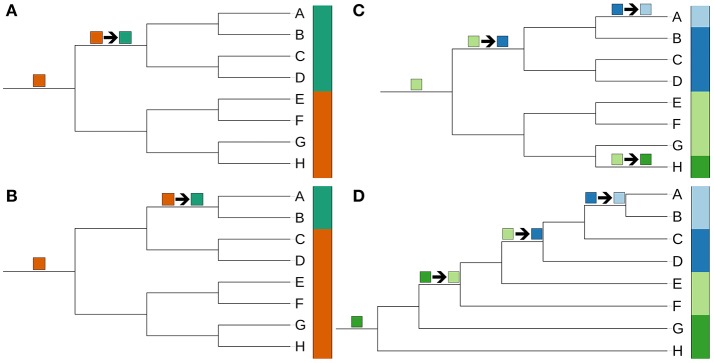
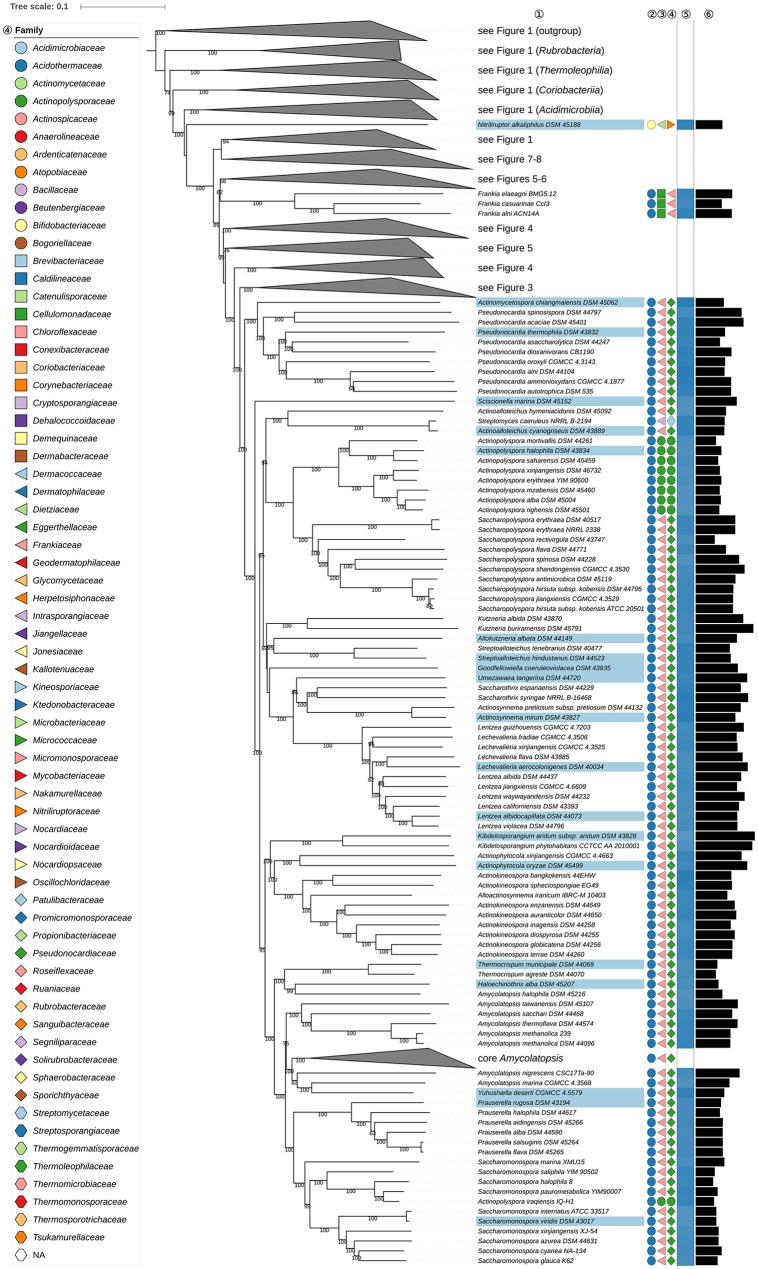
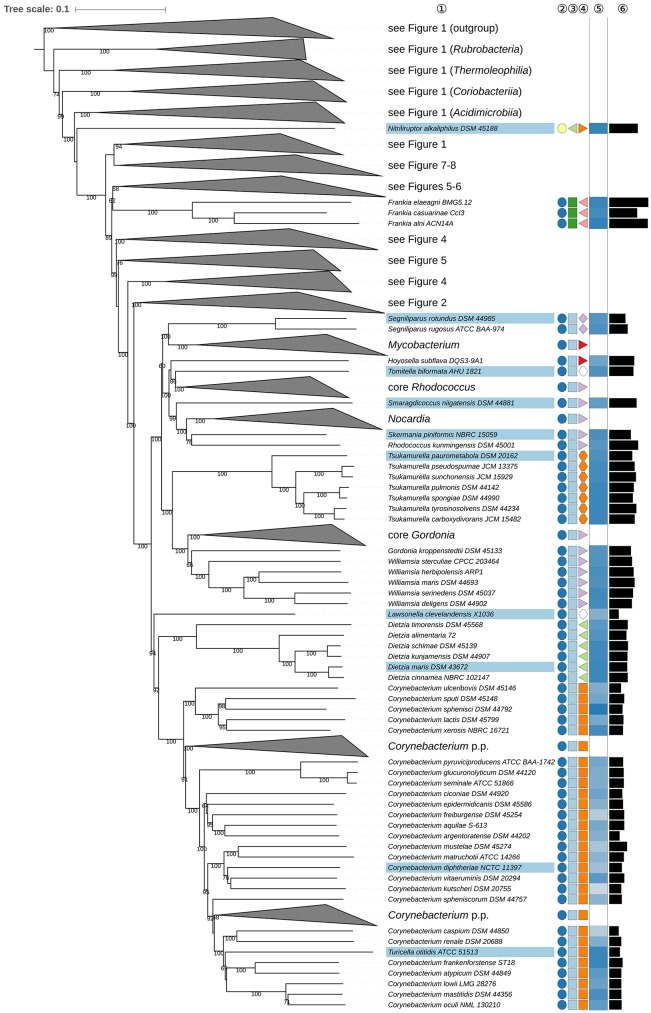
The application of phylogenetic taxonomic procedures led to improvements in the classification of bacteria assigned to the phylum but even so there remains a need to further clarify relationships within a taxon that encompasses organisms of agricultural, biotechnological, clinical, and ecological importance. Classification of the morphologically diverse bacteria belonging to this large phylum based on a limited number of features has proved to be difficult, not least when taxonomic decisions rested heavily on interpretation of poorly resolved 16S rRNA gene trees. Here, draft genome sequences of a large collection of actinobacterial type strains were used to infer phylogenetic trees from genome-scale data using principles drawn from phylogenetic systematics. The majority of taxa were found to be monophyletic but several orders, families, and genera, as well as many species and a few subspecies were shown to be in need of revision leading to proposals for the recognition of 2 orders, 10 families, and 17 genera, as well as the transfer of over 100 species to other genera. In addition, emended descriptions are given for many species mainly involving the addition of data on genome size and DNA G+C content, the former can be considered to be a valuable taxonomic marker in actinobacterial systematics. Many of the incongruities detected when the results of the present study were compared with existing classifications had been recognized from 16S rRNA gene trees though whole-genome phylogenies proved to be much better resolved. The few significant incongruities found between 16S/23S rRNA and whole genome trees underline the pitfalls inherent in phylogenies based upon single gene sequences. Similarly good congruence was found between the discontinuous distribution of phenotypic properties and taxa delineated in the phylogenetic trees though diverse non-monophyletic taxa appeared to be based on the use of plesiomorphic character states as diagnostic features.
系统发育分类学方法的应用改进了属于该门细菌的分类,但即便如此,对于一个包含具有农业、生物技术、临床和生态重要性的生物体的分类单元,仍有必要进一步厘清其内部的关系。基于有限数量的特征对属于这个大门类的形态多样的细菌进行分类已证明是困难的,尤其是当分类学决策严重依赖于对解析度差的16S rRNA基因树的解释时。在这里,利用从系统发育系统学中得出的原理,使用大量放线菌模式菌株的基因组草图序列从基因组规模数据推断系统发育树。发现大多数分类单元是单系的,但有几个目、科和属,以及许多种和一些亚种显示需要修订,从而提出了承认2个目、10个科和17个属的建议,以及将100多个种转移到其他属的提议。此外,还对许多种给出了修订后的描述,主要涉及增加基因组大小和DNA G+C含量的数据,前者可被视为放线菌系统学中有价值的分类标记。当将本研究结果与现有分类进行比较时检测到的许多不一致性在16S rRNA基因树中已被认识到,尽管全基因组系统发育树被证明解析度要好得多。在16S/23S rRNA和全基因组树之间发现的少数显著不一致性突显了基于单基因序列的系统发育中固有的陷阱。同样,在表型特征的不连续分布与系统发育树中划定的分类单元之间也发现了良好的一致性,尽管不同的非单系分类单元似乎是基于使用近祖性状状态作为诊断特征。