Princess Margaret Cancer Centre/University Health Network, Toronto, Ontario, Canada.
Institute of Digestive Disease and Department of Medicine and Therapeutics, State Key Laboratory of Digestive Disease, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong.
PLoS Biol. 2018 Sep 13;16(9):e2006092. doi: 10.1371/journal.pbio.2006092. eCollection 2018 Sep.
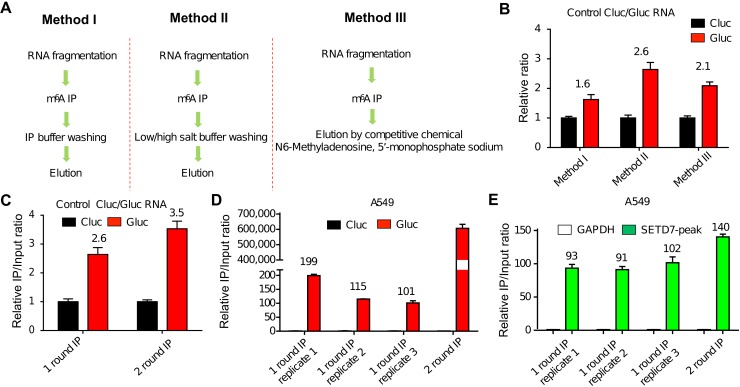
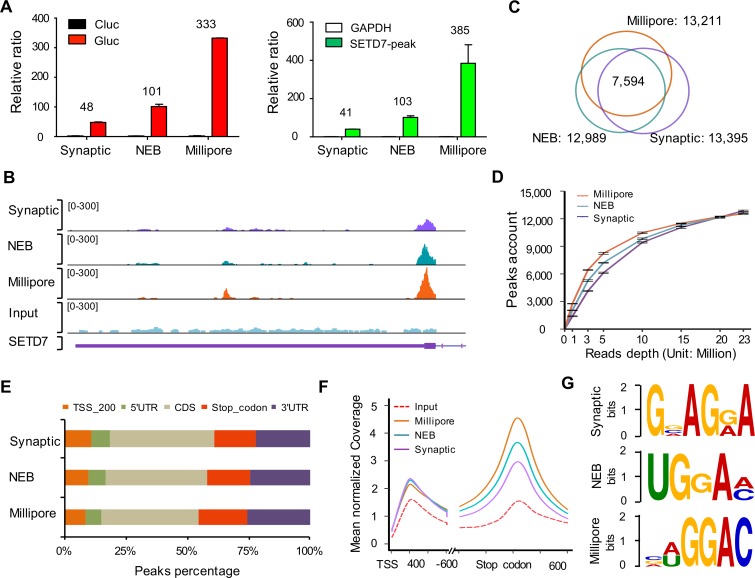
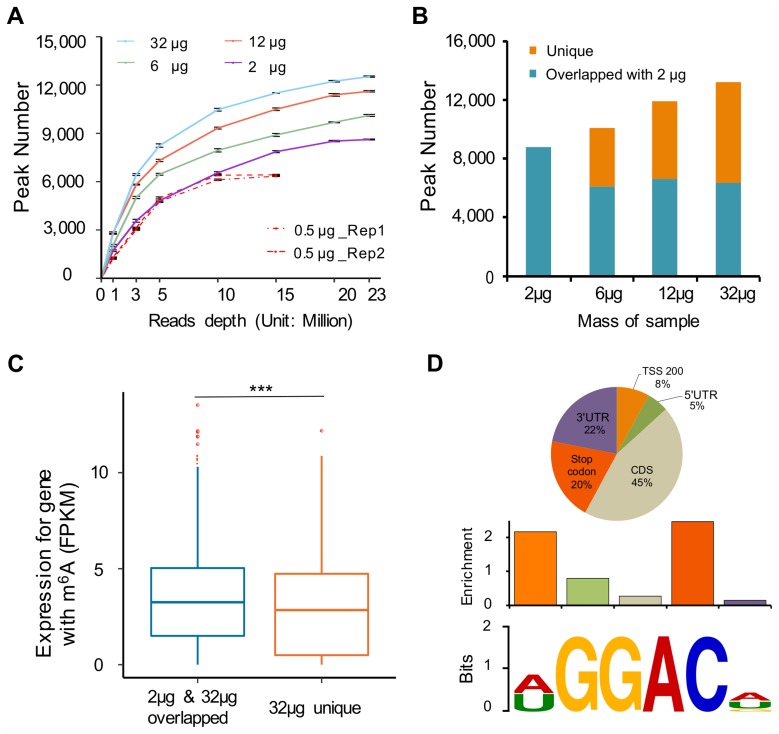
N6-Methyladenosine (m6A) accounts for approximately 0.2% to 0.6% of all adenosine in mammalian mRNA, representing the most abundant internal mRNA modifications. m6A RNA immunoprecipitation followed by high-throughput sequencing (MeRIP-seq) is a powerful technique to map the m6A location transcriptome-wide. However, this method typically requires 300 μg of total RNA, which limits its application to patient tumors. In this study, we present a refined m6A MeRIP-seq protocol and analysis pipeline that can be applied to profile low-input RNA samples from patient tumors. We optimized the key parameters of m6A MeRIP-seq, including the starting amount of RNA, RNA fragmentation, antibody selection, MeRIP washing/elution conditions, methods for RNA library construction, and the bioinformatics analysis pipeline. With the optimized immunoprecipitation (IP) conditions and a postamplification rRNA depletion strategy, we were able to profile the m6A epitranscriptome using 500 ng of total RNA. We identified approximately 12,000 m6A peaks with a high signal-to-noise (S/N) ratio from 2 lung adenocarcinoma (ADC) patient tumors. Through integrative analysis of the transcriptome, m6A epitranscriptome, and proteome data in the same patient tumors, we identified dynamics at the m6A level that account for the discordance between mRNA and protein levels in these tumors. The refined m6A MeRIP-seq method is suitable for m6A epitranscriptome profiling in a limited amount of patient tumors, setting the ground for unraveling the dynamics of the m6A epitranscriptome and the underlying mechanisms in clinical settings.
N6-甲基腺苷(m6A)约占哺乳动物 mRNA 中腺苷的 0.2%至 0.6%,代表最丰富的内部 mRNA 修饰。m6A RNA 免疫沉淀结合高通量测序(MeRIP-seq)是一种在全转录组范围内绘制 m6A 位置的强大技术。然而,该方法通常需要 300μg 的总 RNA,这限制了其在患者肿瘤中的应用。在本研究中,我们提出了一种改进的 m6A MeRIP-seq 方案和分析流程,可应用于分析来自患者肿瘤的低输入 RNA 样本。我们优化了 m6A MeRIP-seq 的关键参数,包括起始 RNA 量、RNA 片段化、抗体选择、MeRIP 洗涤/洗脱条件、RNA 文库构建方法和生物信息学分析流程。通过优化免疫沉淀(IP)条件和扩增后 rRNA 耗竭策略,我们能够使用 500ng 总 RNA 来描绘 m6A 转录组。我们从 2 个肺腺癌(ADC)患者肿瘤中鉴定出约 12000 个具有高信噪比(S/N)的 m6A 峰。通过对同一患者肿瘤中的转录组、m6A 转录组和蛋白质组数据进行综合分析,我们确定了 m6A 水平上的动态变化,这些动态变化解释了这些肿瘤中 mRNA 和蛋白质水平之间的差异。改良的 m6A MeRIP-seq 方法适用于有限量患者肿瘤中的 m6A 转录组描绘,为揭示临床环境中 m6A 转录组的动态变化及其潜在机制奠定了基础。