Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, Henan, China.
McAllister Heart Institute at The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, United States of America.
PLoS Genet. 2018 Sep 17;14(9):e1007664. doi: 10.1371/journal.pgen.1007664. eCollection 2018 Sep.
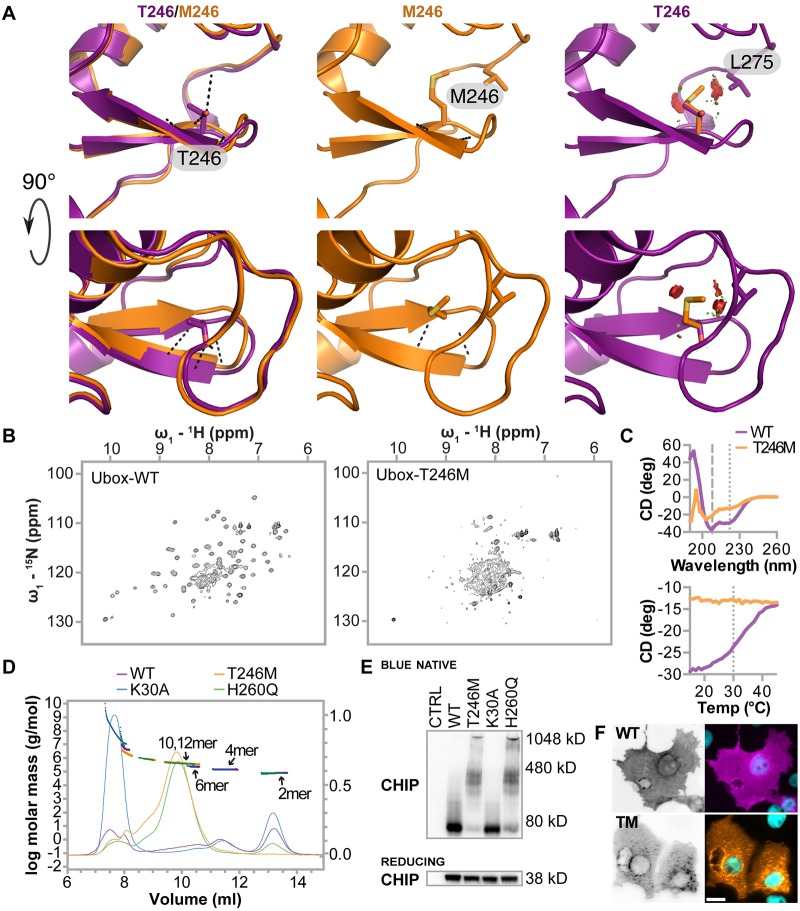
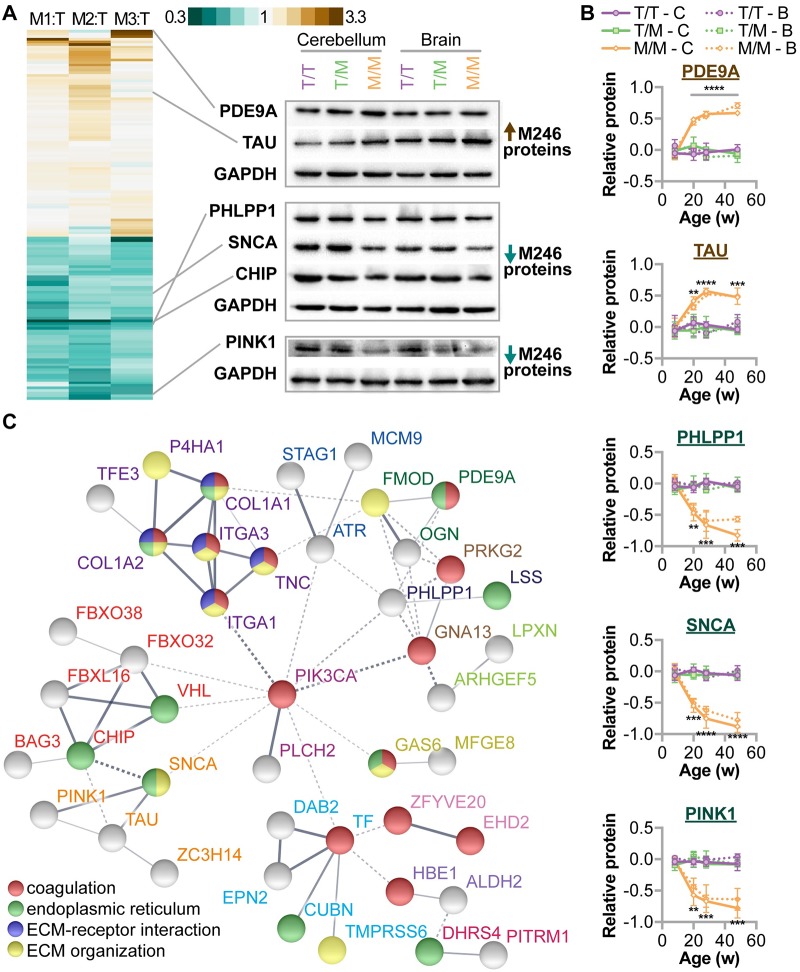
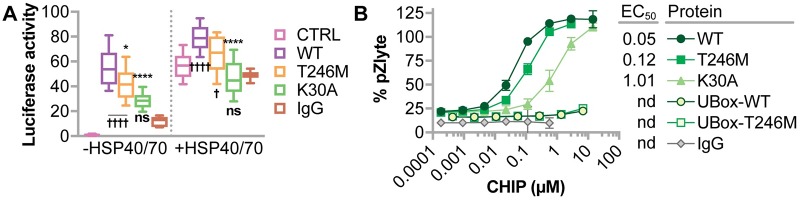
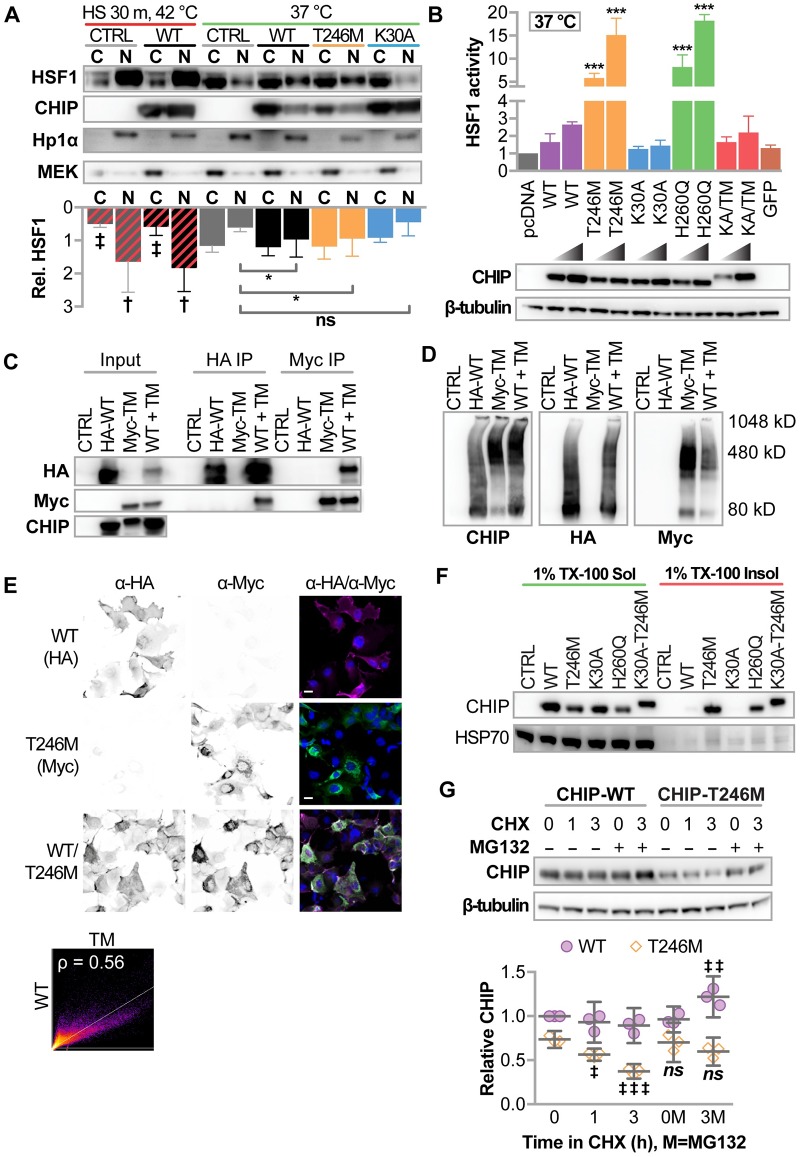
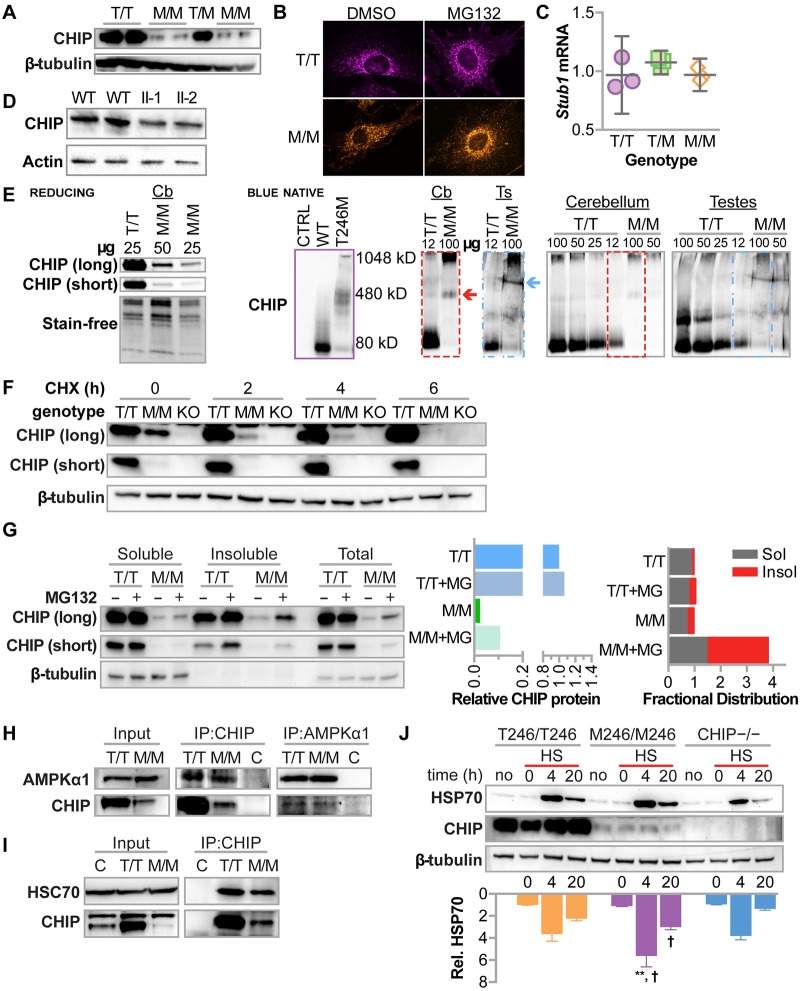
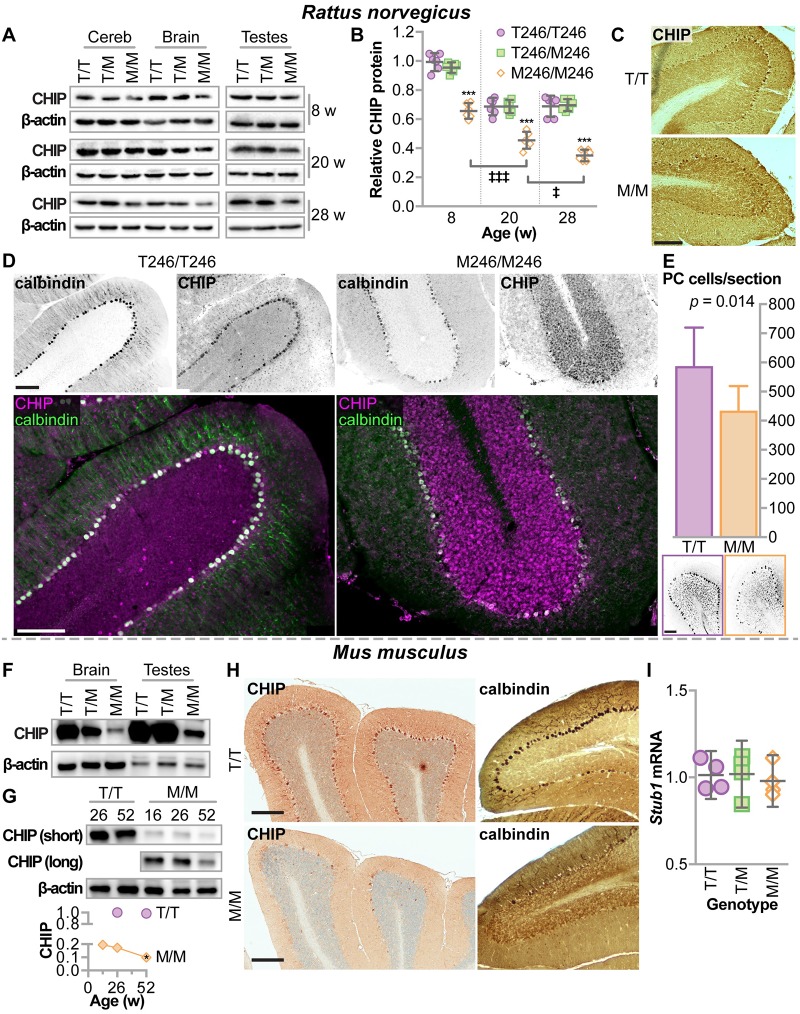
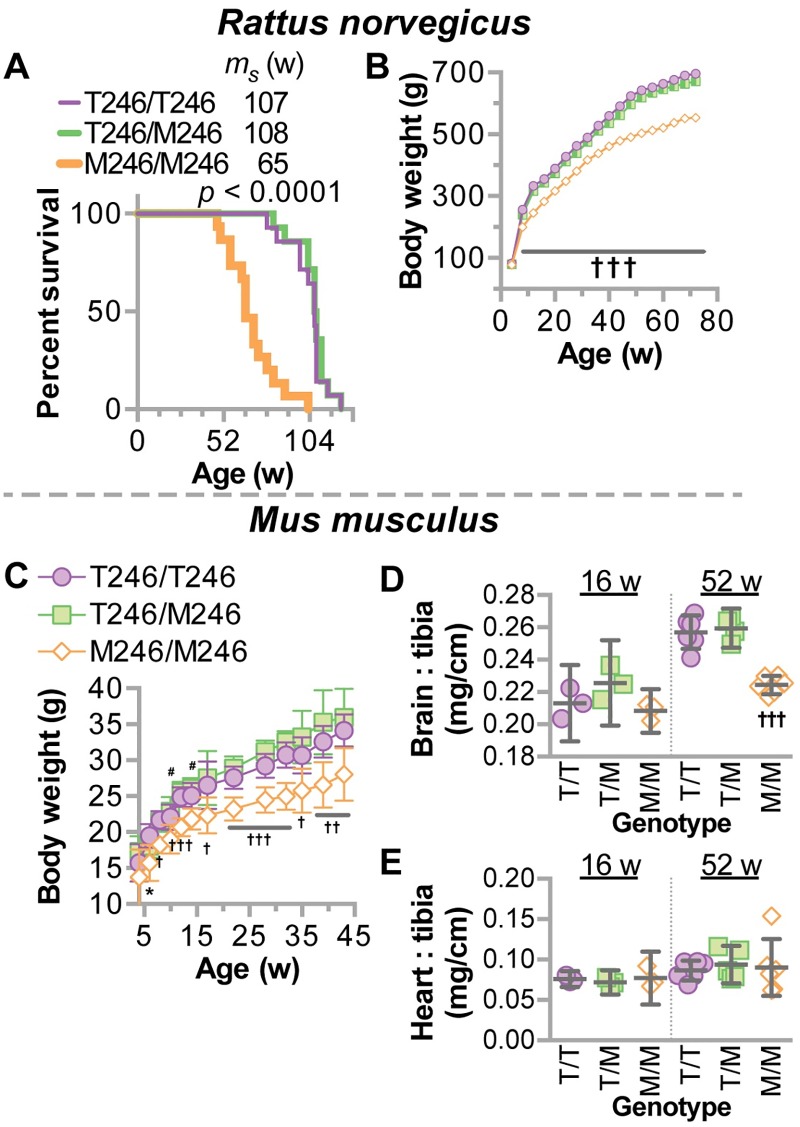
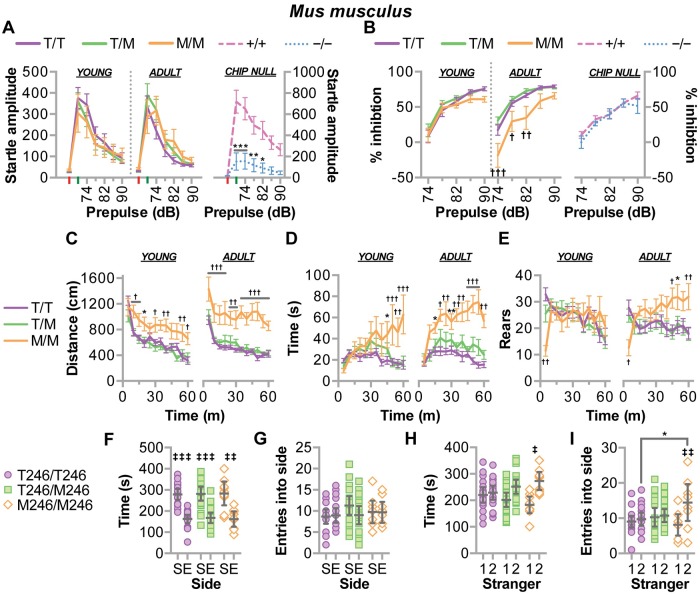
CHIP (carboxyl terminus of heat shock 70-interacting protein) has long been recognized as an active member of the cellular protein quality control system given the ability of CHIP to function as both a co-chaperone and ubiquitin ligase. We discovered a genetic disease, now known as spinocerebellar autosomal recessive 16 (SCAR16), resulting from a coding mutation that caused a loss of CHIP ubiquitin ligase function. The initial mutation describing SCAR16 was a missense mutation in the ubiquitin ligase domain of CHIP (p.T246M). Using multiple biophysical and cellular approaches, we demonstrated that T246M mutation results in structural disorganization and misfolding of the CHIP U-box domain, promoting oligomerization, and increased proteasome-dependent turnover. CHIP-T246M has no ligase activity, but maintains interactions with chaperones and chaperone-related functions. To establish preclinical models of SCAR16, we engineered T246M at the endogenous locus in both mice and rats. Animals homozygous for T246M had both cognitive and motor cerebellar dysfunction distinct from those observed in the CHIP null animal model, as well as deficits in learning and memory, reflective of the cognitive deficits reported in SCAR16 patients. We conclude that the T246M mutation is not equivalent to the total loss of CHIP, supporting the concept that disease-causing CHIP mutations have different biophysical and functional repercussions on CHIP function that may directly correlate to the spectrum of clinical phenotypes observed in SCAR16 patients. Our findings both further expand our basic understanding of CHIP biology and provide meaningful mechanistic insight underlying the molecular drivers of SCAR16 disease pathology, which may be used to inform the development of novel therapeutics for this devastating disease.
CHIP(热休克 70 相互作用蛋白羧基末端)长期以来一直被认为是细胞蛋白质质量控制系统的活跃成员,因为 CHIP 能够作为共伴侣和泛素连接酶发挥作用。我们发现了一种遗传性疾病,现在称为常染色体隐性遗传性小脑共济失调 16 型(SCAR16),其病因是编码突变导致 CHIP 泛素连接酶功能丧失。最初描述 SCAR16 的突变是 CHIP 泛素连接酶结构域中的错义突变(p.T246M)。我们使用多种生物物理和细胞方法证明,T246M 突变导致 CHIP U 盒结构域的结构紊乱和错误折叠,促进寡聚化和增加蛋白酶体依赖性降解。CHIP-T246M 没有连接酶活性,但保持与伴侣蛋白的相互作用和伴侣蛋白相关的功能。为了建立 SCAR16 的临床前模型,我们在小鼠和大鼠的内源性基因座上设计了 T246M。纯合 T246M 的动物既有认知和运动小脑功能障碍,与 CHIP 缺失动物模型观察到的不同,也有学习和记忆缺陷,反映了 SCAR16 患者报告的认知缺陷。我们得出结论,T246M 突变与 CHIP 的完全缺失不同,这支持了这样一种观点,即导致疾病的 CHIP 突变对 CHIP 功能具有不同的生物物理和功能影响,这可能与在 SCAR16 患者中观察到的临床表型谱直接相关。我们的研究结果不仅进一步扩展了我们对 CHIP 生物学的基本认识,还为 SCAR16 疾病病理学的分子驱动因素提供了有意义的机制见解,这可能用于为这种毁灭性疾病开发新的治疗方法。