Medway School of Pharmacy, University of Kent and University of Greenwich, Chatham Maritime, Kent, UK.
Red de Investigación Cardiovascular, Instituto de Salud Carlos III, Madrid, Spain.
J Physiol. 2019 Feb;597(4):1087-1101. doi: 10.1113/JP277275. Epub 2018 Nov 24.
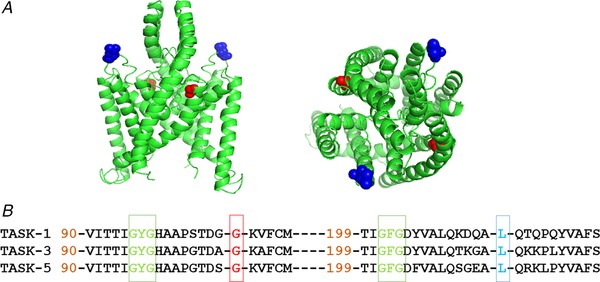
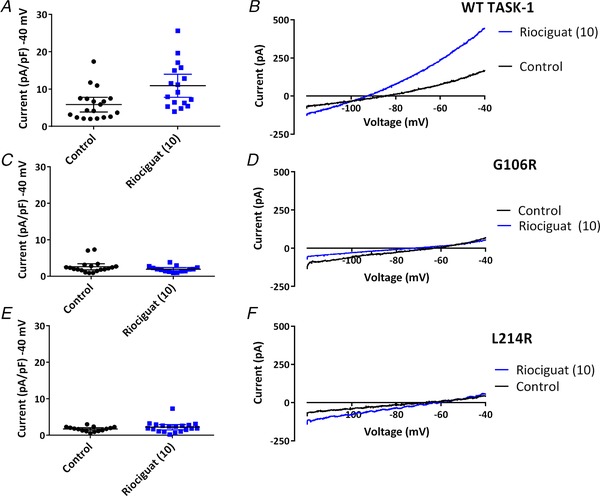
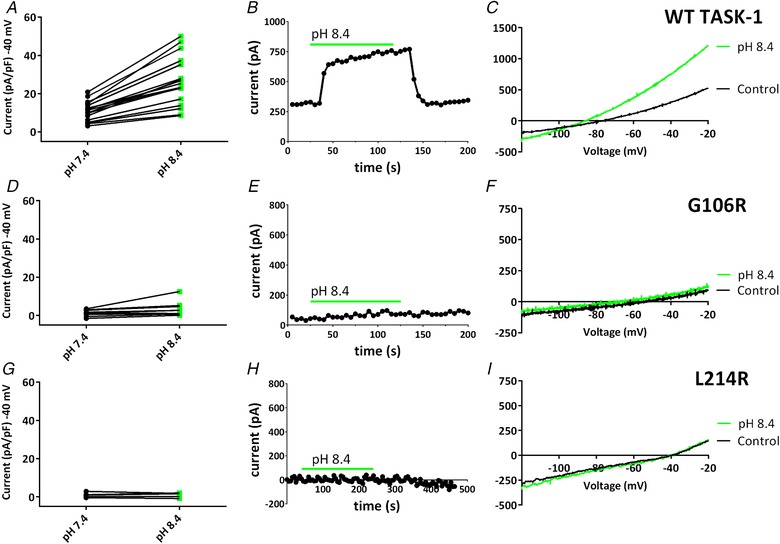
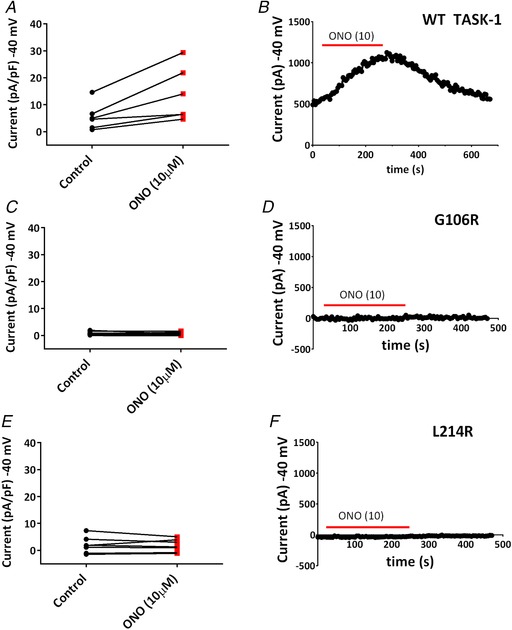
The TASK-1 channel gene (KCNK3) has been identified as a possible disease-causing gene in heritable pulmonary arterial hypertension (PAH). In the present study, we show that novel mutated TASK-1 channels, seen in PAH patients, have a substantially reduced current compared to wild-type TASK-1 channels. These mutated TASK-1 channels are located at the plasma membrane to the same degree as wild-type TASK-1 channels. ONO-RS-082 and alkaline pH 8.4 both activate TASK-1 channels but do not recover current through mutant TASK-1 channels. We show that the guanylate cyclase activator, riociguat, a novel treatment for PAH, enhances current through TASK-1 channels but does not recover current through mutant TASK-1 channels.
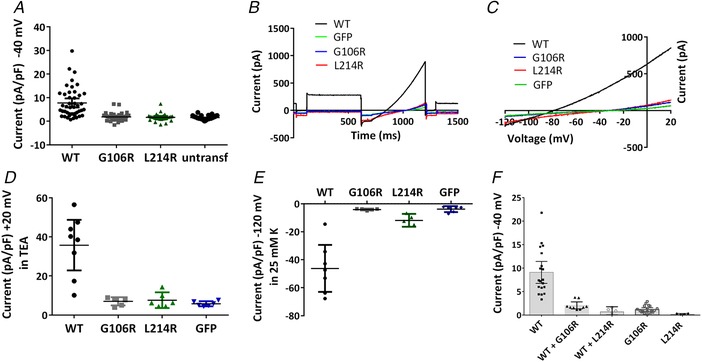
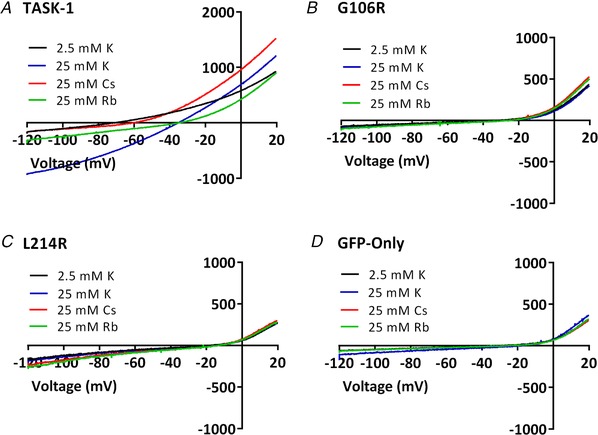
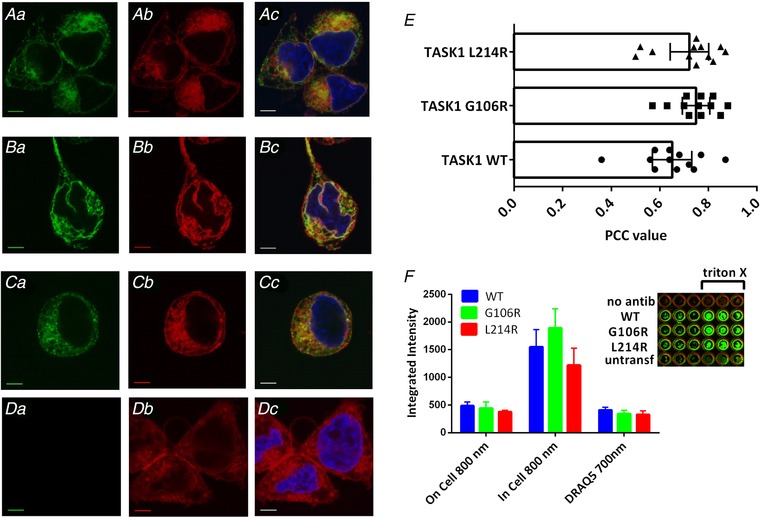
Pulmonary arterial hypertension (PAH) affects ∼15-50 people per million. KCNK3, the gene that encodes the two pore domain potassium channel TASK-1 (K2P3.1), has been identified as a possible disease-causing gene in heritable PAH. Recently, two new mutations have been identified in KCNK3 in PAH patients: G106R and L214R. The present study aimed to characterize the functional properties and regulation of wild-type (WT) and mutated TASK-1 channels and determine how these might contribute to PAH and its treatment. Currents through WT and mutated human TASK-1 channels transiently expressed in tsA201 cells were measured using whole-cell patch clamp electrophysiology. Localization of fluorescence-tagged channels was visualized using confocal microscopy and quantified with in-cell and on-cell westerns. G106R or L214R mutated channels were located at the plasma membrane to the same degree as WT channels; however, their current was markedly reduced compared to WT TASK-1 channels. Functional current through these mutated channels could not be restored using activators of WT TASK-1 channels (pH 8.4, ONO-RS-082). The guanylate cyclase activator, riociguat, enhanced current through WT TASK-1 channels; however, similar to the other activators investigated, riociguat did not have any effect on current through mutated TASK-1 channels. Thus, novel mutations in TASK-1 seen in PAH substantially alter the functional properties of these channels. Current through these channels could not be restored by activators of TASK-1 channels. Riociguat enhancement of current through TASK-1 channels could contribute to its therapeutic benefit in the treatment of PAH.
TASK-1 通道基因(KCNK3)已被确定为遗传性肺动脉高压(PAH)的潜在致病基因。在本研究中,我们发现 PAH 患者中存在的新型突变 TASK-1 通道的电流明显低于野生型 TASK-1 通道。这些突变的 TASK-1 通道位于质膜上的程度与野生型 TASK-1 通道相同。ONO-RS-082 和碱性 pH8.4 均能激活 TASK-1 通道,但不能恢复突变 TASK-1 通道的电流。我们表明,鸟苷酸环化酶激活剂 riociguat 是一种治疗 PAH 的新方法,可增强 TASK-1 通道的电流,但不能恢复突变 TASK-1 通道的电流。
肺动脉高压(PAH)影响每百万人口中约 15-50 人。编码双孔域钾通道 TASK-1(K2P3.1)的 KCNK3 基因已被确定为遗传性 PAH 的潜在致病基因。最近,在 PAH 患者的 KCNK3 中发现了两种新的突变:G106R 和 L214R。本研究旨在描述野生型(WT)和突变 TASK-1 通道的功能特性和调节,并确定这些特性如何导致 PAH 及其治疗。使用全细胞膜片钳电生理学测量瞬时表达在 tsA201 细胞中的 WT 和突变人 TASK-1 通道的电流。使用共焦显微镜可视化荧光标记通道的定位,并使用细胞内和细胞外western blot 进行量化。与 WT 通道一样,G106R 或 L214R 突变通道定位于质膜,但与 WT TASK-1 通道相比,其电流明显降低。使用 WT TASK-1 通道的激活剂(pH8.4、ONO-RS-082)不能恢复这些突变通道的功能电流。鸟苷酸环化酶激活剂 riociguat 增强了 WT TASK-1 通道的电流;然而,与研究的其他激活剂一样,riociguat 对突变 TASK-1 通道的电流没有任何影响。因此,PAH 中 TASK-1 的新型突变极大地改变了这些通道的功能特性。TASK-1 通道的激活剂不能恢复这些通道的电流。Riociguat 增强 TASK-1 通道的电流可能有助于其在治疗 PAH 中的治疗益处。