Department of Computer Science and Engineering, National Taiwan Ocean University, Keelung, Taiwan.
Department of Computer Science and Information Engineering, National Taipei University of Technology, Taipei, Taiwan.
BMC Bioinformatics. 2018 Aug 13;19(Suppl 9):284. doi: 10.1186/s12859-018-2278-z.
Transcriptomic sequencing (RNA-seq) related applications allow for rapid explorations due to their high-throughput and relatively fast experimental capabilities, providing unprecedented progress in gene functional annotation, gene regulation analysis, and environmental factor verification. However, with increasing amounts of sequenced reads and reference model species, the selection of appropriate reference species for gene annotation has become a new challenge.
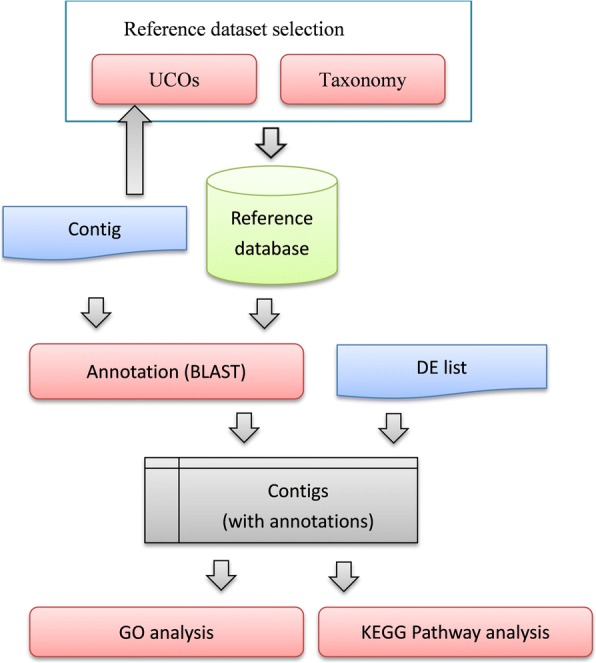
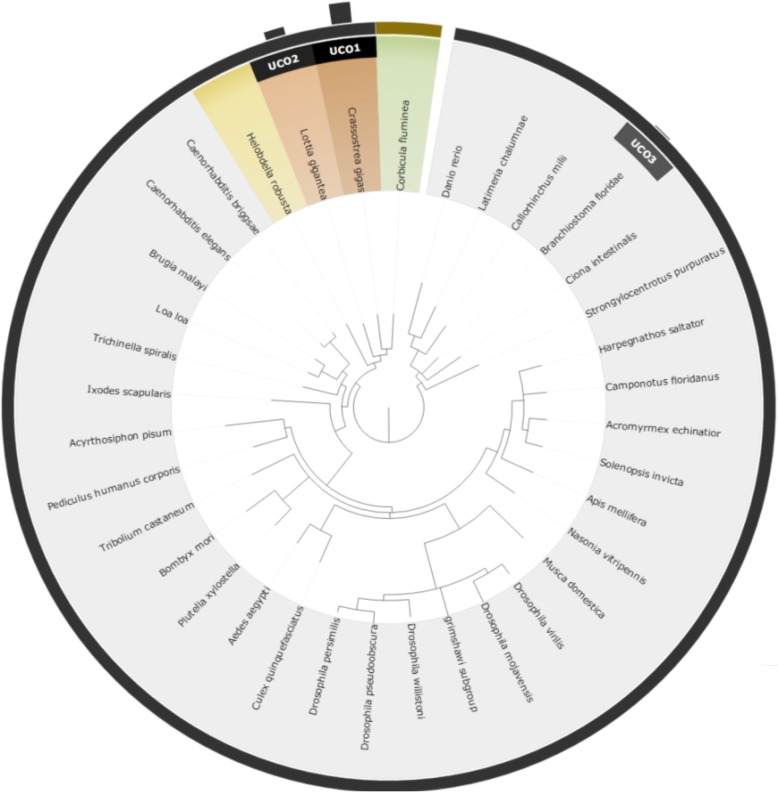
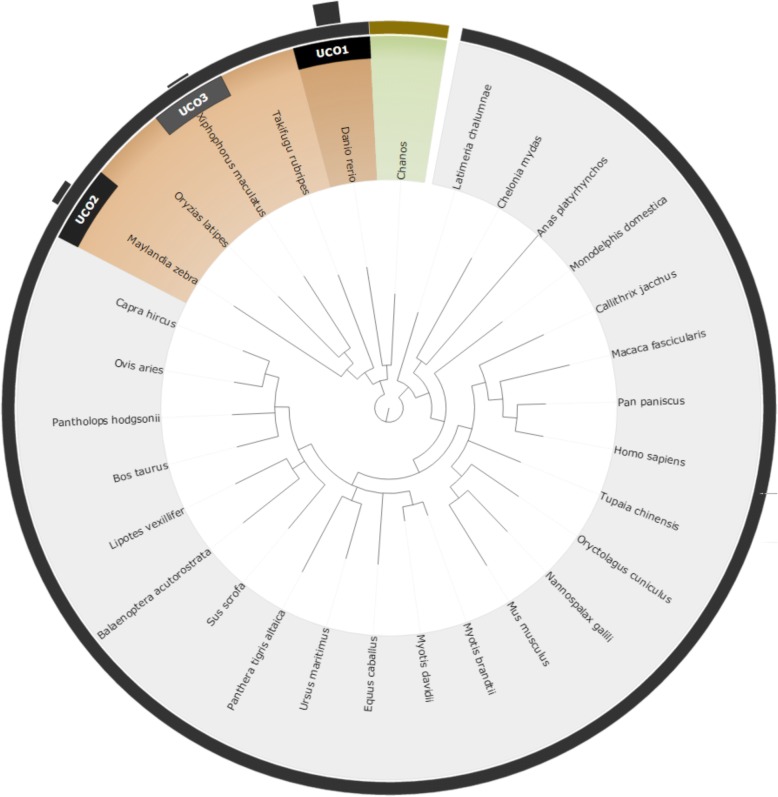
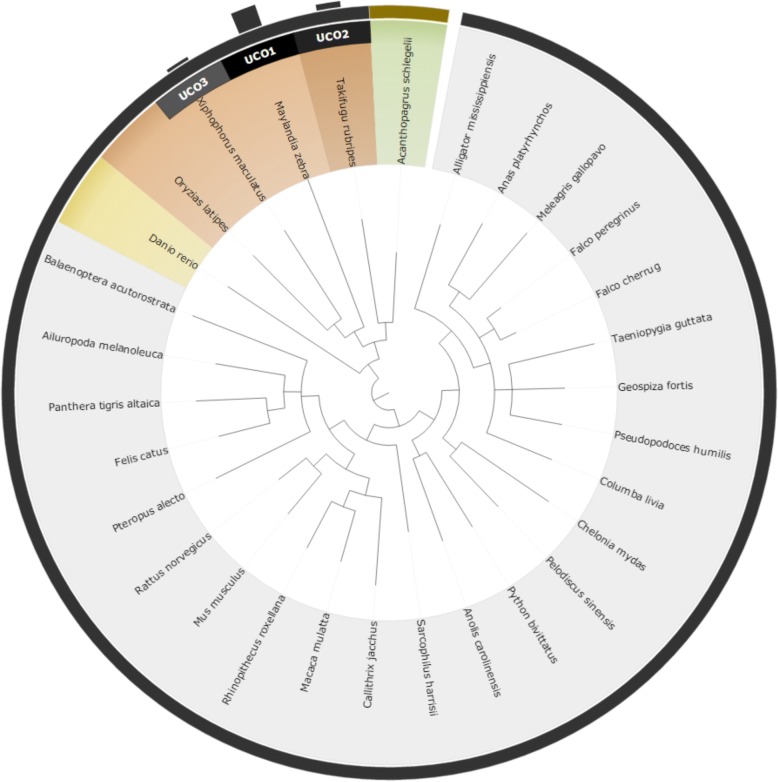
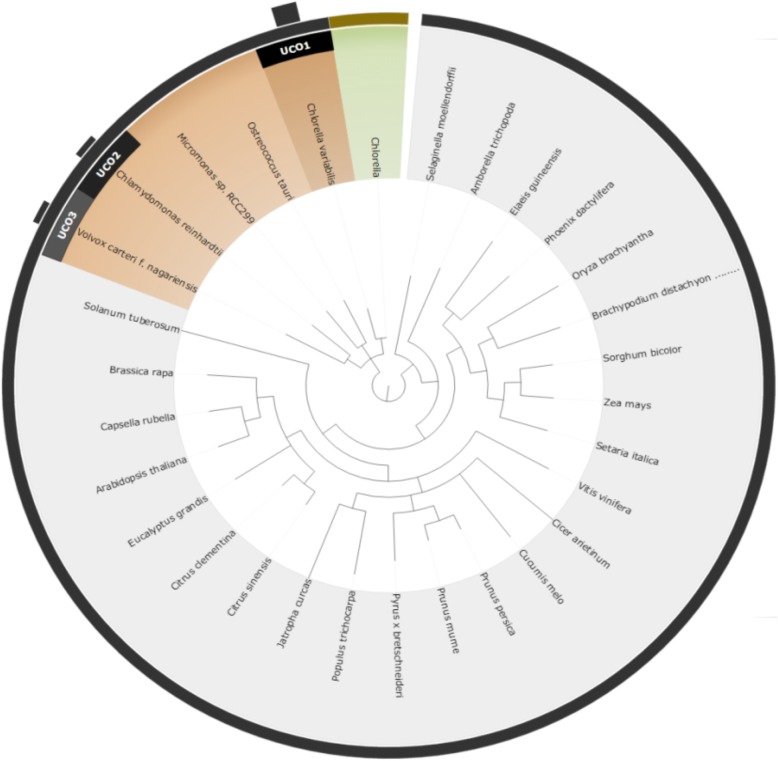
We proposed a novel approach for finding the most effective reference model species through taxonomic associations and ultra-conserved orthologous (UCO) gene comparisons among species. An online system for multiple species selection (MSS) for RNA-seq differential expression analysis was developed, and comprehensive genomic annotations from 291 reference model eukaryotic species were retrieved from the RefSeq, KEGG, and UniProt databases.
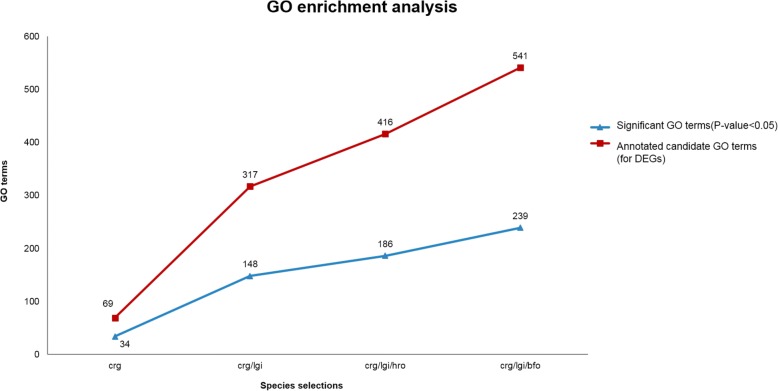
Using the proposed MSS pipeline, gene ontology and biological pathway enrichment analysis can be efficiently achieved, especially in the case of transcriptomic analysis of non-model organisms. The results showed that the proposed method solved problems related to limitations in annotation information and provided a roughly twenty-fold reduction in computational time, resulting in more accurate results than those of traditional approaches of using a single model reference species or the large non-redundant reference database.
Selection of appropriate reference model species helps to reduce missing annotation information, allowing for more comprehensive results than those obtained with a single model reference species. In addition, adequate model species selection reduces the computational time significantly while retaining the same order of accuracy. The proposed system indeed provides superior performance by selecting appropriate multiple species for transcriptomic analysis compared to traditional approaches.
转录组测序(RNA-seq)相关应用因其高通量和相对较快的实验能力而能够快速探索,为基因功能注释、基因调控分析和环境因素验证提供了前所未有的进展。然而,随着测序读段和参考模型物种数量的增加,为基因注释选择合适的参考物种已成为新的挑战。
我们提出了一种通过分类群关联和物种间超保守直系同源(UCO)基因比较来寻找最有效参考模型物种的新方法。开发了一个用于 RNA-seq 差异表达分析的多物种选择(MSS)在线系统,并从 RefSeq、KEGG 和 UniProt 数据库中检索了 291 个参考真核模型物种的综合基因组注释。
使用所提出的 MSS 管道,可以有效地进行基因本体论和生物途径富集分析,特别是在非模型生物的转录组分析中。结果表明,该方法解决了注释信息有限的问题,并将计算时间减少了约二十倍,与使用单个模型参考物种或大型非冗余参考数据库的传统方法相比,结果更准确。
选择合适的参考模型物种有助于减少缺失的注释信息,提供比使用单个模型参考物种更全面的结果。此外,适当的模型物种选择可以大大减少计算时间,同时保持相同的准确性。与传统方法相比,该系统通过为转录组分析选择合适的多个物种,确实提供了卓越的性能。