Department of Computer Science, University of Saskatchewan, Saskatoon, S7N 5C9, Canada.
Augmented Intelligence & Precision Health Laboratory (AIPHL), Research Institute of the McGill University Health Centre, Montreal, H4A 3S5, Canada.
BMC Bioinformatics. 2021 Mar 16;22(1):125. doi: 10.1186/s12859-021-04055-1.
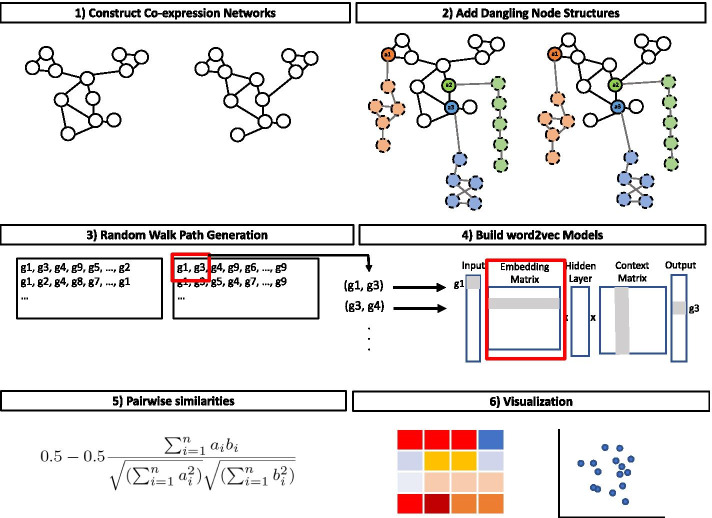
Gene co-expression networks (GCNs) are not easily comparable due to their complex structure. In this paper, we propose a tool, Juxtapose, together with similarity measures that can be utilized for comparative transcriptomics between a set of organisms. While we focus on its application to comparing co-expression networks across species in evolutionary studies, Juxtapose is also generalizable to co-expression network comparisons across tissues or conditions within the same species.
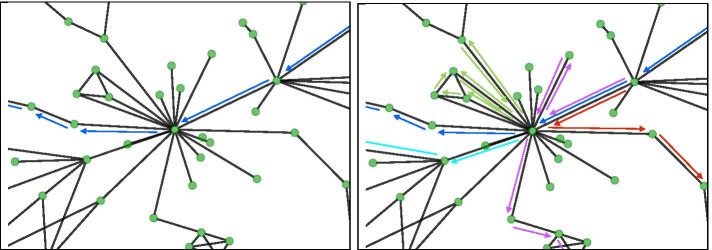
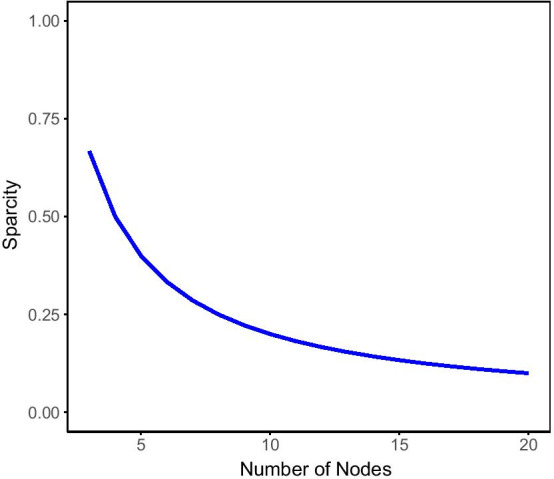
A word embedding strategy commonly used in natural language processing was utilized in order to generate gene embeddings based on walks made throughout the GCNs. Juxtapose was evaluated based on its ability to embed the nodes of synthetic structures in the networks consistently while also generating biologically informative results. Evaluation of the techniques proposed in this research utilized RNA-seq datasets from GTEx, a multi-species experiment of prefrontal cortex samples from the Gene Expression Omnibus, as well as synthesized datasets. Biological evaluation was performed using gene set enrichment analysis and known gene relationships in literature.
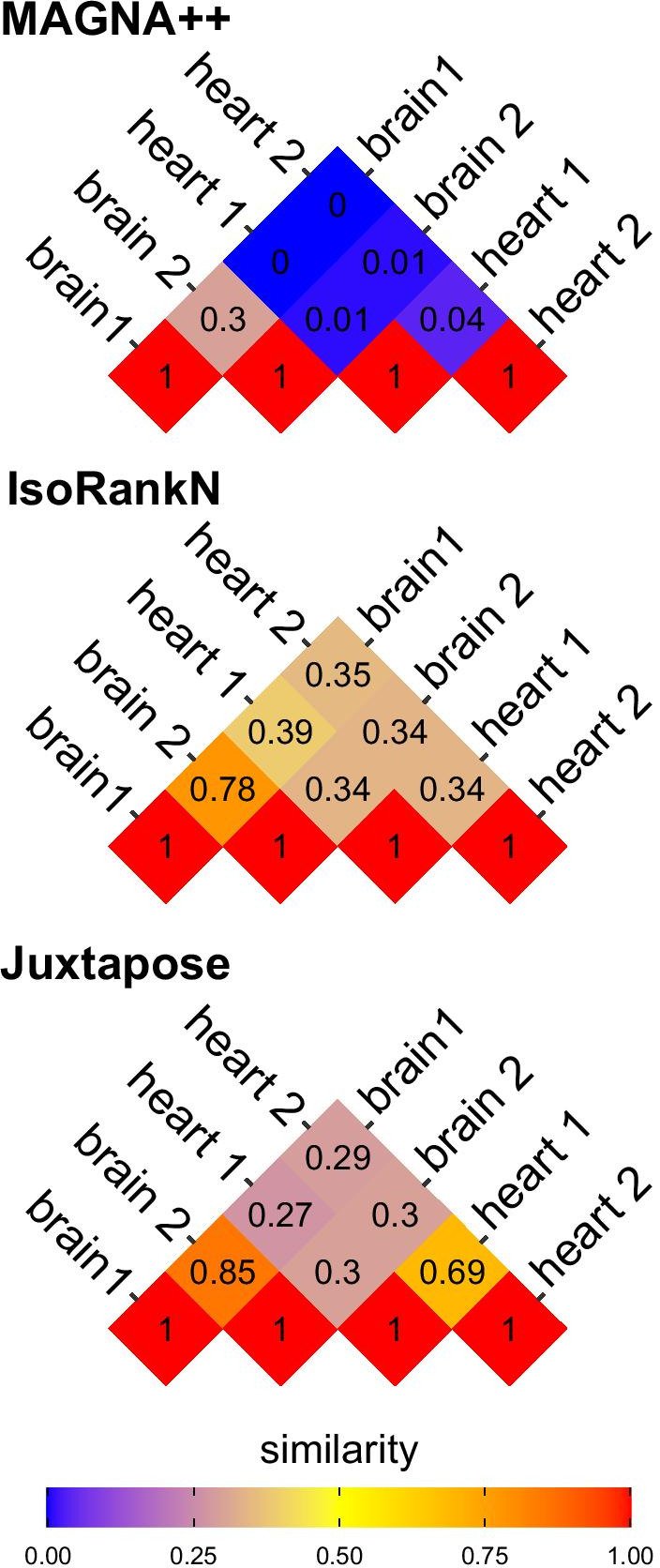
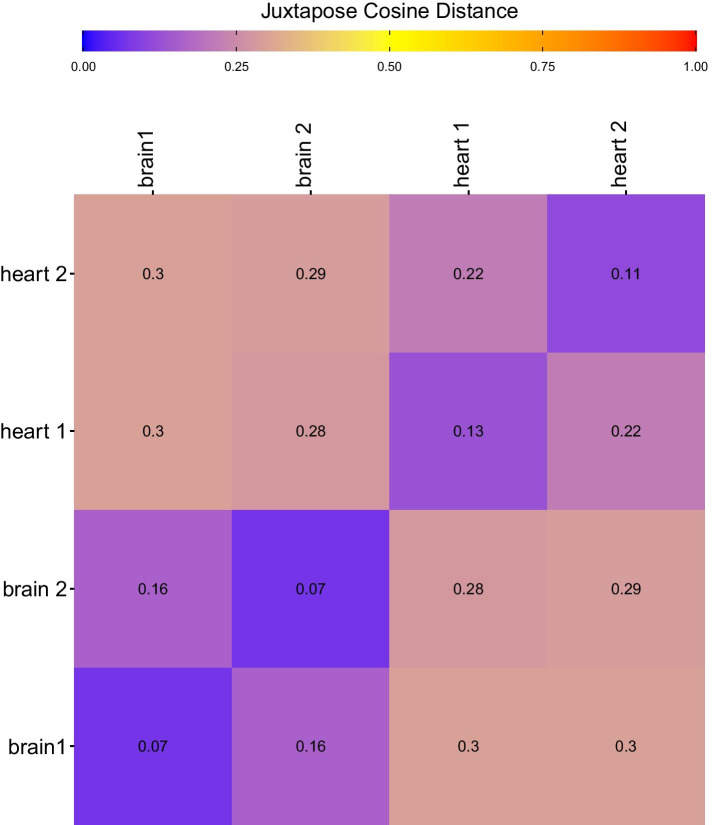
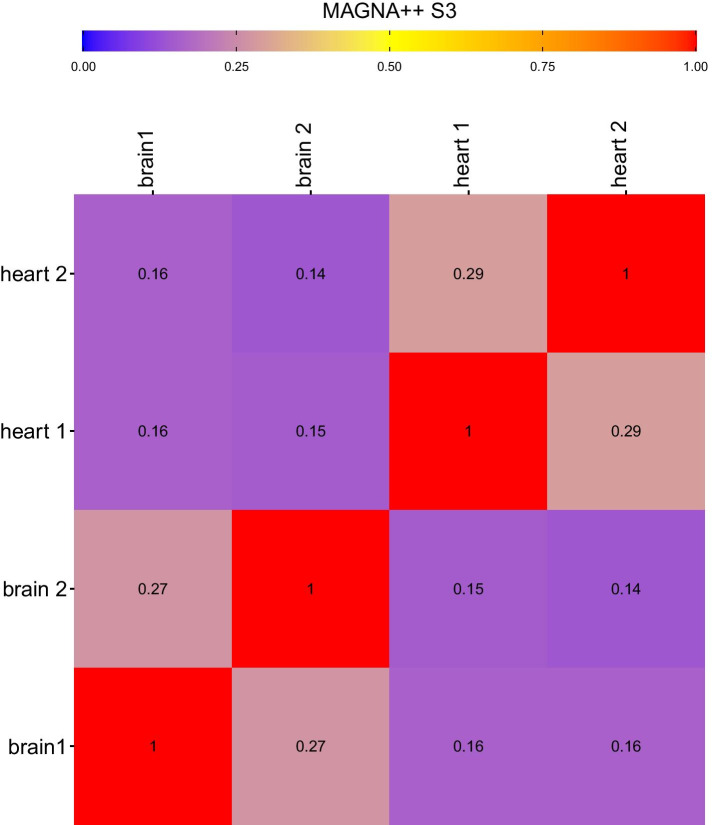
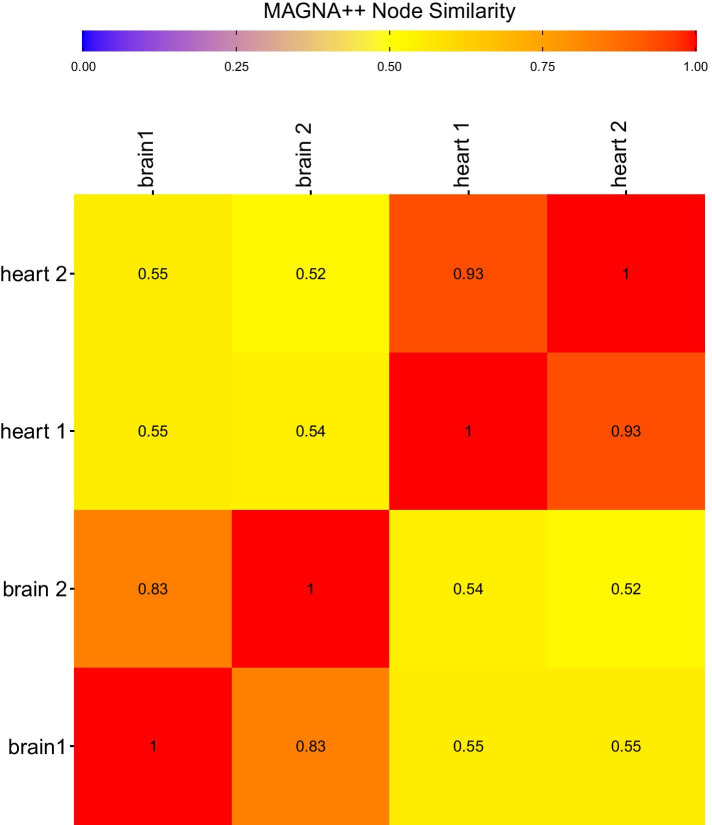
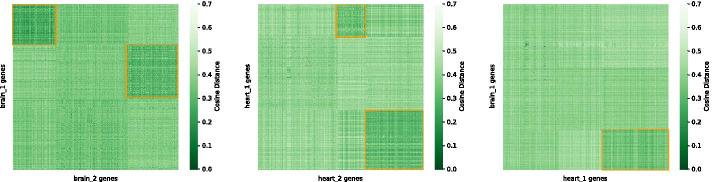
We show that Juxtapose is capable of globally aligning synthesized networks as well as identifying areas that are conserved in real gene co-expression networks without reliance on external biological information. Furthermore, output from a matching algorithm that uses cosine distance between GCN embeddings is shown to be an informative measure of similarity that reflects the amount of topological similarity between networks.
Juxtapose can be used to align GCNs without relying on known biological similarities and enables post-hoc analyses using biological parameters, such as orthology of genes, or conserved or variable pathways.
A development version of the software used in this paper is available at https://github.com/klovens/juxtapose.
由于基因共表达网络(GCN)的结构复杂,它们不容易进行比较。在本文中,我们提出了一个名为 Juxtapose 的工具以及一些相似性度量标准,可用于在一组生物体之间进行比较转录组学。虽然我们专注于将其应用于进化研究中比较物种间的共表达网络,但 Juxtapose 也可推广到同一物种内比较组织或条件的共表达网络。
我们利用自然语言处理中常用的词嵌入策略,根据在 GCN 中进行的游走生成基因嵌入。根据其在一致嵌入网络中合成结构节点的能力以及生成具有生物学意义的结果的能力来评估 Juxtapose。本研究中提出的技术的评估利用了 GTEx 的 RNA-seq 数据集,该数据集来自 Gene Expression Omnibus 的前额叶皮质样本的多物种实验,以及合成数据集。使用基因集富集分析和文献中的已知基因关系进行生物学评估。
我们表明,Juxtapose 能够全局对齐合成网络,并识别真实基因共表达网络中保守的区域,而无需依赖外部生物学信息。此外,使用 GCN 嵌入之间余弦距离的匹配算法的输出被证明是一种信息量的相似性度量,反映了网络之间拓扑相似性的程度。
Juxtapose 可以在不依赖已知生物学相似性的情况下对齐 GCN,并支持使用生物学参数(例如基因的同源性)或保守或可变途径进行事后分析。
本文中使用的软件的开发版本可在 https://github.com/klovens/juxtapose 上获得。