Department of Biomedical Informatics, Harvard Medical School, 10 Shattuck Street, Boston, 02115, MA, USA.
Department of Computer Science and Engineering, University of Connecticut, 371 Fairfield Way, Unit 2155, Storrs, 06269, CT, USA.
BMC Genomics. 2018 Aug 13;19(Suppl 6):566. doi: 10.1186/s12864-018-4920-6.
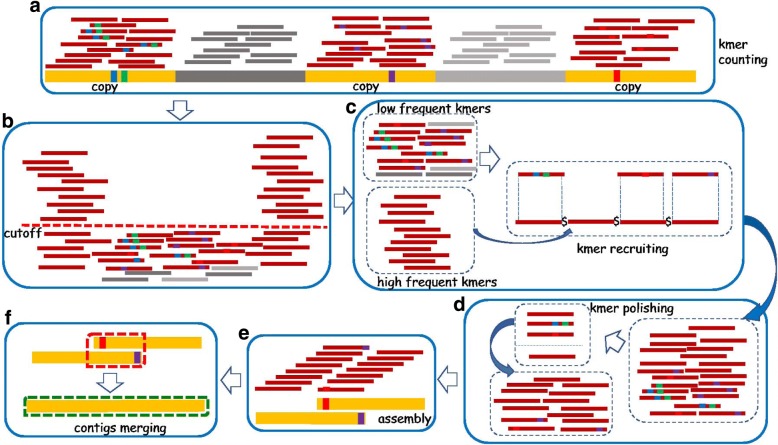
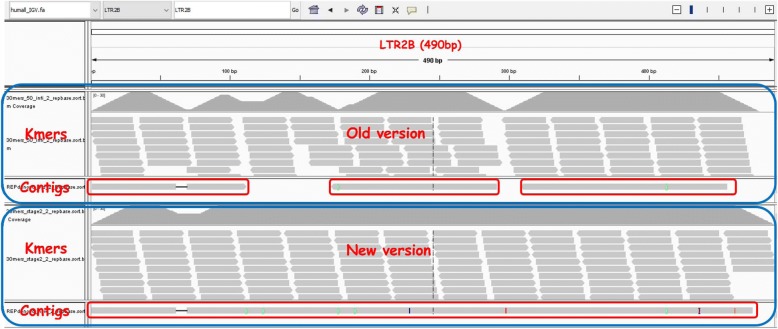
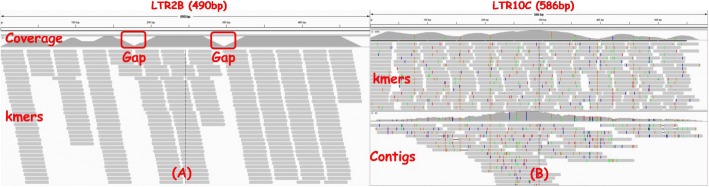
Repeat elements are important components of most eukaryotic genomes. Most existing tools for repeat analysis rely either on high quality reference genomes or existing repeat libraries. Thus, it is still challenging to do repeat analysis for species with highly repetitive or complex genomes which often do not have good reference genomes or annotated repeat libraries. Recently we developed a computational method called REPdenovo that constructs consensus repeat sequences directly from short sequence reads, which outperforms an existing tool called RepARK. One major issue with REPdenovo is that it doesn't perform well for repeats with relatively high divergence rates or low copy numbers. In this paper, we present an improved approach for constructing consensus repeats directly from short reads. Comparing with the original REPdenovo, the improved approach uses more repeat-related k-mers and improves repeat assembly quality using a consensus-based k-mer processing method.
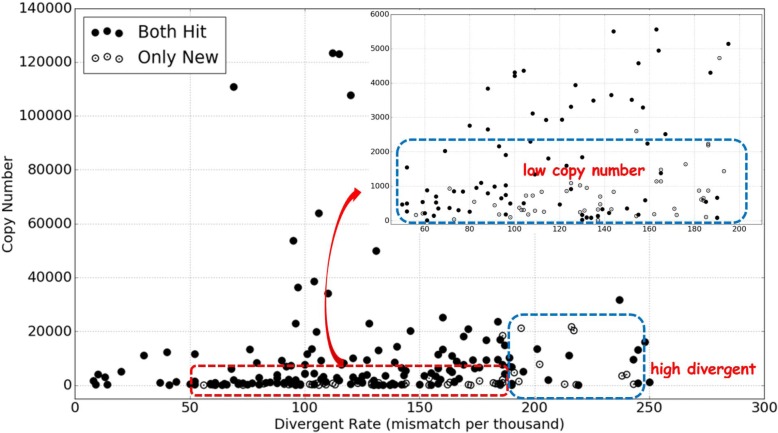
We compare the performance of the new method with REPdenovo and RepARK on Human, Arabidopsis thaliana and Drosophila melanogaster short sequencing data. And the new method fully constructs more repeats in Repbase than the original REPdenovo and RepARK, especially for repeats of higher divergence rates and lower copy number. We also apply our new method on Hummingbird data which doesn't have a known repeat library, and it constructs many repeat elements that can be validated using PacBio long reads.
We propose an improved method for reconstructing repeat elements directly from short sequence reads. The results show that our new method can assemble more complete repeats than REPdenovo (and also RepARK). Our new approach has been implemented as part of the REPdenovo software package, which is available for download at https://github.com/Reedwarbler/REPdenovo .
重复元件是大多数真核生物基因组的重要组成部分。大多数现有的重复分析工具要么依赖于高质量的参考基因组,要么依赖于现有的重复文库。因此,对于具有高度重复或复杂基因组的物种,进行重复分析仍然具有挑战性,这些物种通常没有良好的参考基因组或注释的重复文库。最近,我们开发了一种称为 REPdenovo 的计算方法,该方法可以直接从短序列读取构建一致的重复序列,其性能优于称为 RepARK 的现有工具。REPdenovo 的一个主要问题是,对于具有相对较高分歧率或低拷贝数的重复,其性能不佳。在本文中,我们提出了一种从短读取直接构建一致重复的改进方法。与原始的 REPdenovo 相比,改进的方法使用了更多与重复相关的 k-mer,并使用基于一致的 k-mer 处理方法来提高重复组装质量。
我们将新方法与 REPdenovo 和 RepARK 在人类、拟南芥和果蝇短测序数据上的性能进行了比较。与原始的 REPdenovo 和 RepARK 相比,新方法在 Repbase 中完全构建了更多的重复,尤其是对于分歧率较高和拷贝数较低的重复。我们还将我们的新方法应用于 Hummingbird 数据,该数据没有已知的重复文库,它构建了许多可以使用 PacBio 长读取进行验证的重复元素。
我们提出了一种从短序列读取直接重建重复元件的改进方法。结果表明,我们的新方法可以比 REPdenovo(和 RepARK)组装更完整的重复。我们的新方法已作为 REPdenovo 软件包的一部分实现,可在 https://github.com/Reedwarbler/REPdenovo 上下载。