Bakhtiarizadeh Mohammad Reza, Hosseinpour Batool, Shahhoseini Maryam, Korte Arthur, Gifani Peyman
Department of Animal and Poultry Science, College of Aburaihan, University of Tehran, Tehran, Iran.
Department of Agriculture, Iranian Research Organization for Science and Technology, Tehran, Iran.
Front Genet. 2018 Oct 12;9:453. doi: 10.3389/fgene.2018.00453. eCollection 2018.
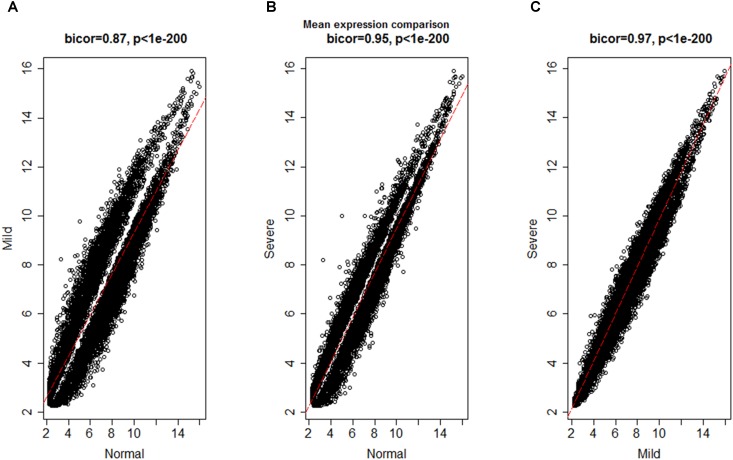
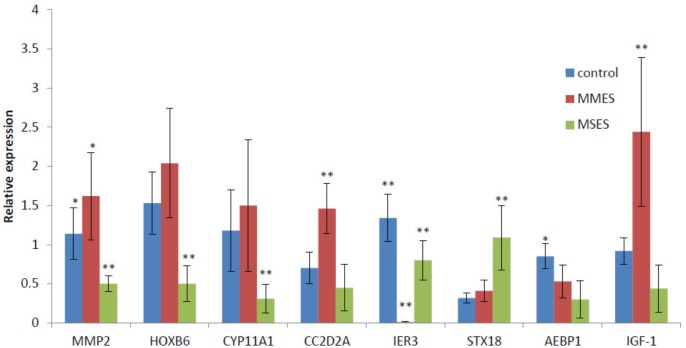
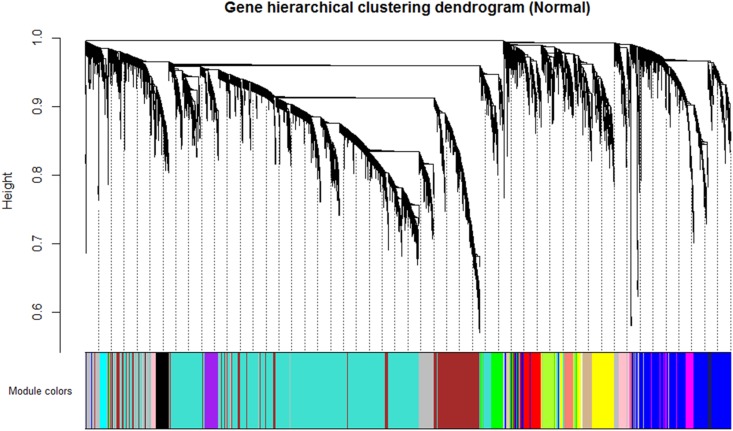
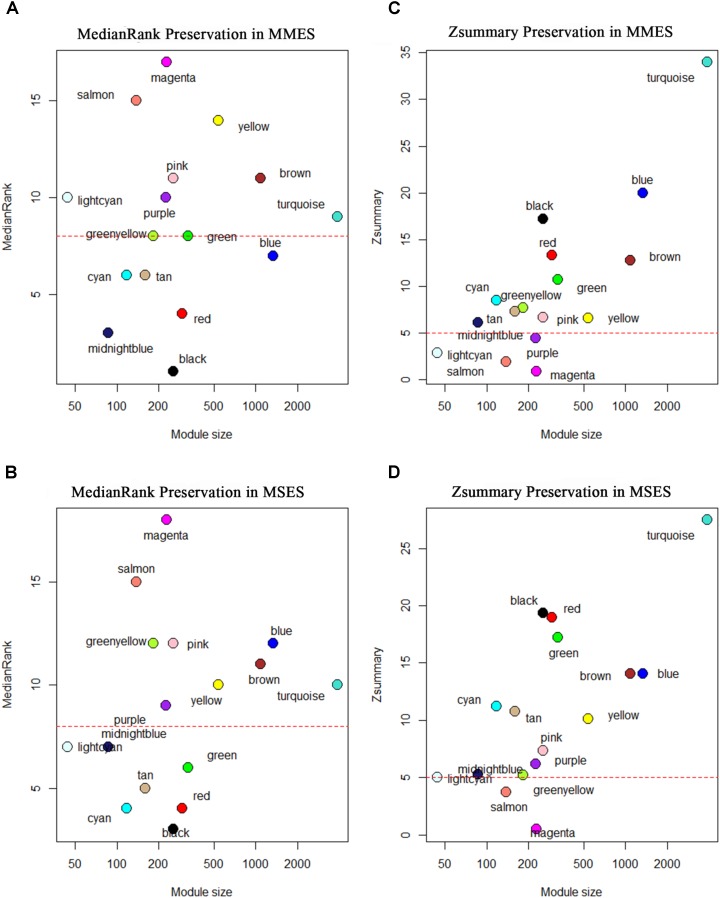
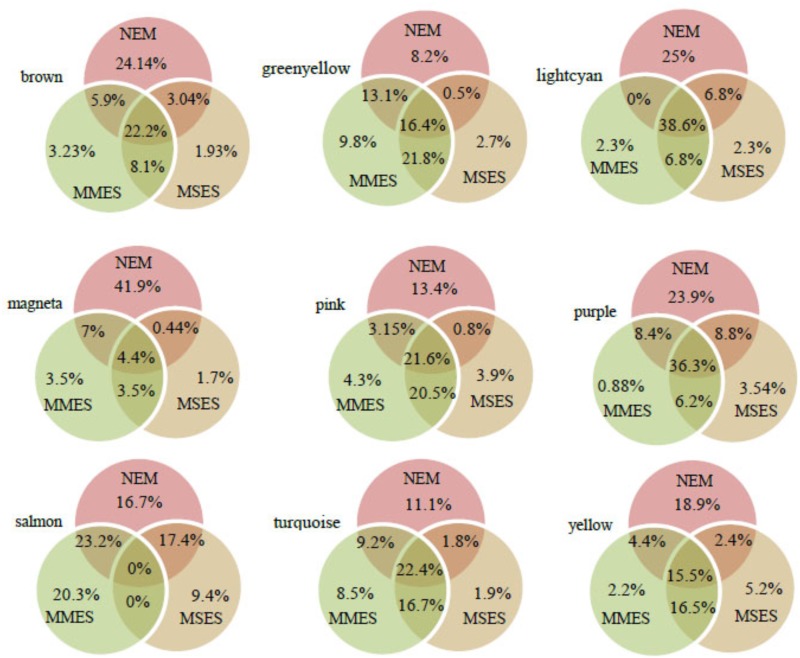
Although many genes have been identified using high throughput technologies in endometriosis (ES), only a small number of individual genes have been analyzed functionally. This is due to the complexity of the disease that has different stages and is affected by various genetic and environmental factors. Many genes are upregulated or downregulated at each stage of the disease, thus making it difficult to identify key genes. In addition, little is known about the differences between the different stages of the disease. We assumed that the study of the identified genes in ES at a system-level can help to better understand the molecular mechanism of the disease at different stages of the development. We used publicly available microarray data containing archived endometrial samples from women with minimal/mild endometriosis (MMES), mild/severe endometriosis (MSES) and without endometriosis. Using weighted gene co-expression analysis (WGCNA), functional modules were derived from normal endometrium (NEM) as the reference sample. Subsequently, we tested whether the topology or connectivity pattern of the modules was preserved in MMES and/or MSES. Common and specific hub genes were identified in non-preserved modules. Accordingly, hub genes were detected in the non-preserved modules at each stage. We identified sixteen co-expression modules. Of the 16 modules, nine were non-preserved in both MMES and MSES whereas five were preserved in NEM, MMES, and MSES. Importantly, two non-preserved modules were found in either MMES or MSES, highlighting differences between the two stages of the disease. Analyzing the hub genes in the non-preserved modules showed that they mostly lost or gained their centrality in NEM after developing the disease into MMES and MSES. The same scenario was observed, when the severeness of the disease switched from MMES to MSES. Interestingly, the expression analysis of the new selected gene candidates including CC2D2A, AEBP1, HOXB6, IER3, and STX18 as well as IGF-1, CYP11A1 and MMP-2 could validate such shifts between different stages. The overrepresented gene ontology (GO) terms were enriched in specific modules, such as genetic disposition, estrogen dependence, progesterone resistance and inflammation, which are known as endometriosis hallmarks. Some modules uncovered novel co-expressed gene clusters that were not previously discovered.
尽管利用高通量技术已在子宫内膜异位症(ES)中鉴定出许多基因,但只有少数单个基因进行了功能分析。这是由于该疾病的复杂性,它具有不同阶段且受多种遗传和环境因素影响。许多基因在疾病的每个阶段都会上调或下调,因此难以鉴定关键基因。此外,对于该疾病不同阶段之间的差异了解甚少。我们假设在系统水平上研究ES中已鉴定的基因有助于更好地理解疾病在不同发育阶段的分子机制。我们使用了公开可用的微阵列数据,这些数据包含来自轻度/轻微子宫内膜异位症(MMES)、轻度/重度子宫内膜异位症(MSES)以及无子宫内膜异位症女性的存档子宫内膜样本。使用加权基因共表达分析(WGCNA),以正常子宫内膜(NEM)作为参考样本得出功能模块。随后,我们测试这些模块的拓扑结构或连接模式在MMES和/或MSES中是否得以保留。在未保留的模块中鉴定出共同的和特定的枢纽基因。相应地,在每个阶段的未保留模块中检测到枢纽基因。我们鉴定出16个共表达模块。在这16个模块中,9个在MMES和MSES中均未保留,而5个在NEM、MMES和MSES中均得以保留。重要的是,在MMES或MSES中发现了两个未保留的模块,突出了该疾病两个阶段之间的差异。对未保留模块中的枢纽基因进行分析表明,在疾病发展为MMES和MSES后,它们大多在NEM中失去或获得了其中心地位。当疾病严重程度从MMES转变为MSES时也观察到了同样的情况。有趣的是,对新选择的基因候选物(包括CC2D2A、AEBP1、HOXB6、IER3和STX18以及IGF-1、CYP11A1和MMP-2)的表达分析可以验证不同阶段之间的这种变化。过度富集的基因本体(GO)术语在特定模块中富集,例如遗传易感性、雌激素依赖性、孕激素抵抗和炎症,这些都是已知的子宫内膜异位症特征。一些模块揭示了以前未发现的新的共表达基因簇。