University of Melbourne Department of Surgery, St. Vincent's Hospital, Melbourne, VIC, 3065, Australia.
Division of Bioinformatics, Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, 3051, Australia.
BMC Bioinformatics. 2018 Nov 6;19(1):404. doi: 10.1186/s12859-018-2435-4.
Gene set scoring provides a useful approach for quantifying concordance between sample transcriptomes and selected molecular signatures. Most methods use information from all samples to score an individual sample, leading to unstable scores in small data sets and introducing biases from sample composition (e.g. varying numbers of samples for different cancer subtypes). To address these issues, we have developed a truly single sample scoring method, and associated R/Bioconductor package singscore ( https://bioconductor.org/packages/singscore ).
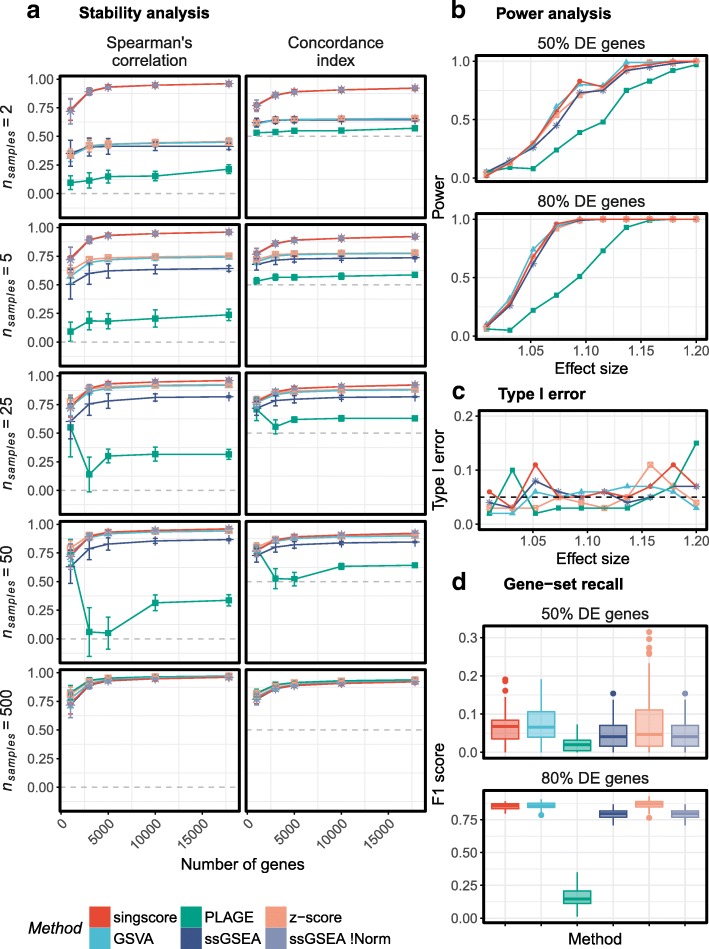
We use multiple cancer data sets to compare singscore against widely-used methods, including GSVA, z-score, PLAGE, and ssGSEA. Our approach does not depend upon background samples and scores are thus stable regardless of the composition and number of samples being scored. In contrast, scores obtained by GSVA, z-score, PLAGE and ssGSEA can be unstable when less data are available (N < 25). The singscore method performs as well as the best performing methods in terms of power, recall, false positive rate and computational time, and provides consistently high and balanced performance across all these criteria. To enhance the impact and utility of our method, we have also included a set of functions implementing visual analysis and diagnostics to support the exploration of molecular phenotypes in single samples and across populations of data.
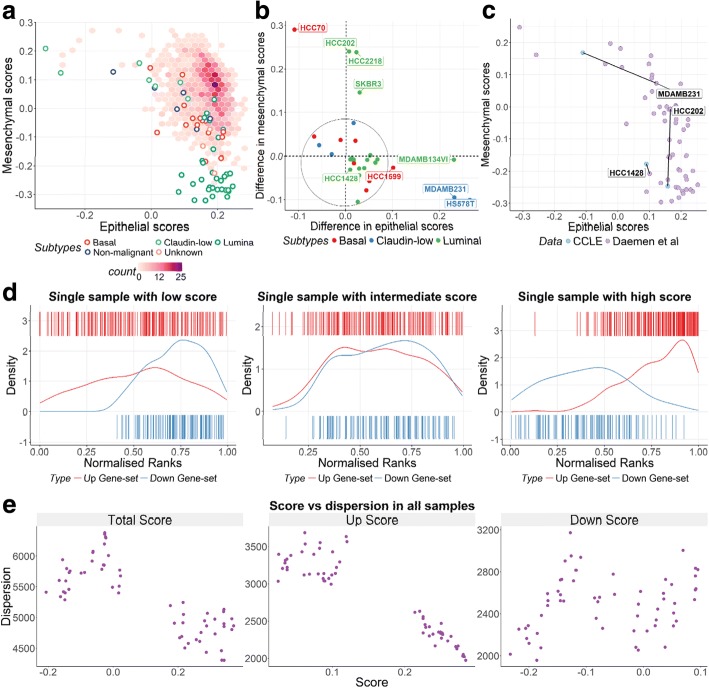
The singscore method described here functions independent of sample composition in gene expression data and thus it provides stable scores, which are particularly useful for small data sets or data integration. Singscore performs well across all performance criteria, and includes a suite of powerful visualization functions to assist in the interpretation of results. This method performs as well as or better than other scoring approaches in terms of its power to distinguish samples with distinct biology and its ability to call true differential gene sets between two conditions. These scores can be used for dimensional reduction of transcriptomic data and the phenotypic landscapes obtained by scoring samples against multiple molecular signatures may provide insights for sample stratification.
基因集评分提供了一种有用的方法,可以量化样本转录组与选定分子特征之间的一致性。大多数方法使用所有样本的信息来对单个样本进行评分,这导致在小数据集和样本组成(例如不同癌症亚型的样本数量不同)中引入偏差的评分不稳定。为了解决这些问题,我们开发了一种真正的单个样本评分方法,并开发了相关的 R/Bioconductor 包 singscore(https://bioconductor.org/packages/singscore)。
我们使用多个癌症数据集来比较 singscore 与广泛使用的方法,包括 GSVA、z 分数、PLAGE 和 ssGSEA。我们的方法不依赖于背景样本,因此评分是稳定的,与评分样本的组成和数量无关。相比之下,当数据较少(N<25)时,GSVA、z 分数、PLAGE 和 ssGSEA 获得的评分可能不稳定。singscore 方法在功效、召回率、假阳性率和计算时间方面与表现最好的方法一样好,并在所有这些标准上提供一致的高且平衡的性能。为了增强我们方法的影响和实用性,我们还包括了一组实现可视化分析和诊断的功能,以支持在单个样本和数据群体中探索分子表型。
这里描述的 singscore 方法独立于基因表达数据中的样本组成,因此它提供稳定的评分,这对于小数据集或数据集成特别有用。singscore 在所有性能标准上表现良好,并包括一套强大的可视化功能,以帮助解释结果。就区分具有不同生物学特性的样本的能力及其在两种情况下调用真实差异基因集的能力而言,该方法的性能与其他评分方法一样好或更好。这些分数可用于转录组数据的降维,并且对样本与多个分子特征进行评分获得的表型景观可能为样本分层提供见解。