Faculty of Veterinary Medicine, Rajamangala University of Technology Tawan-ok, Chonburi, Thailand.
School of Veterinary Medicine, Faculty of Health and Medical Sciences, University of Surrey, Surrey, UK.
BMC Genomics. 2018 Nov 6;19(1):801. doi: 10.1186/s12864-018-5137-4.
Salmonella enterica is a significant foodborne pathogen, which can be transmitted via several distinct routes, and reports on acquisition of antimicrobial resistance (AMR) are increasing. To better understand the association between human Salmonella clinical isolates and the potential environmental/animal reservoirs, whole genome sequencing (WGS) was used to investigate the epidemiology and AMR patterns within Salmonella isolates from two adjacent US states.
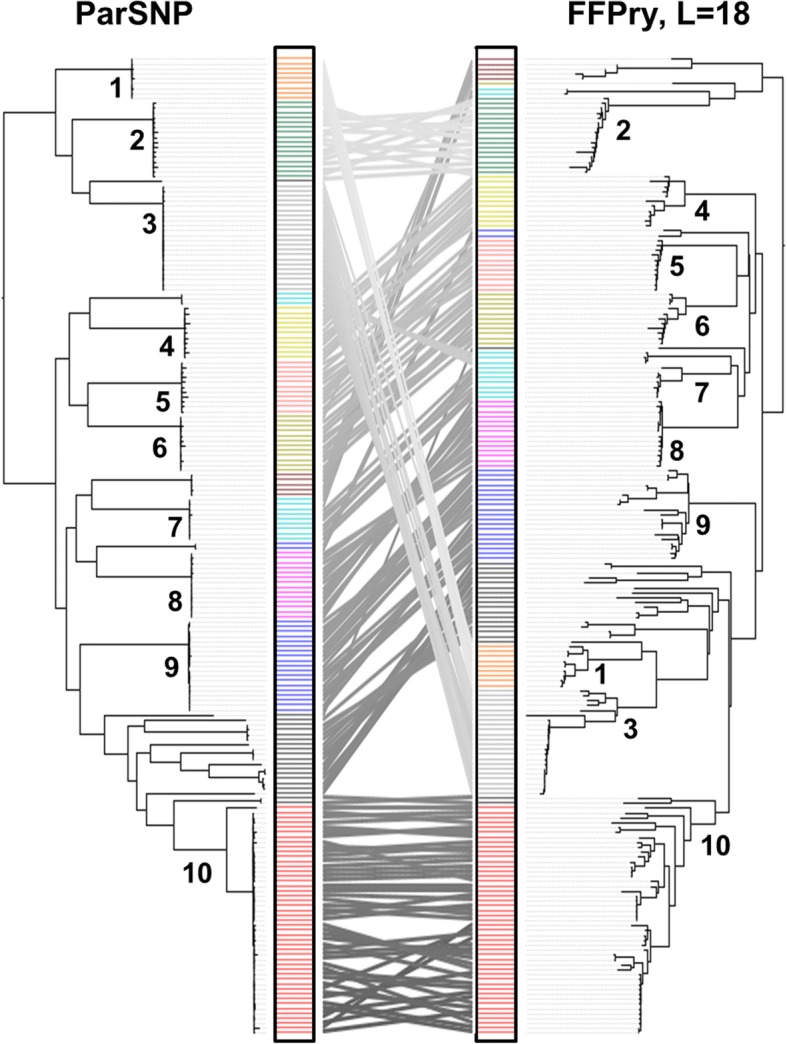
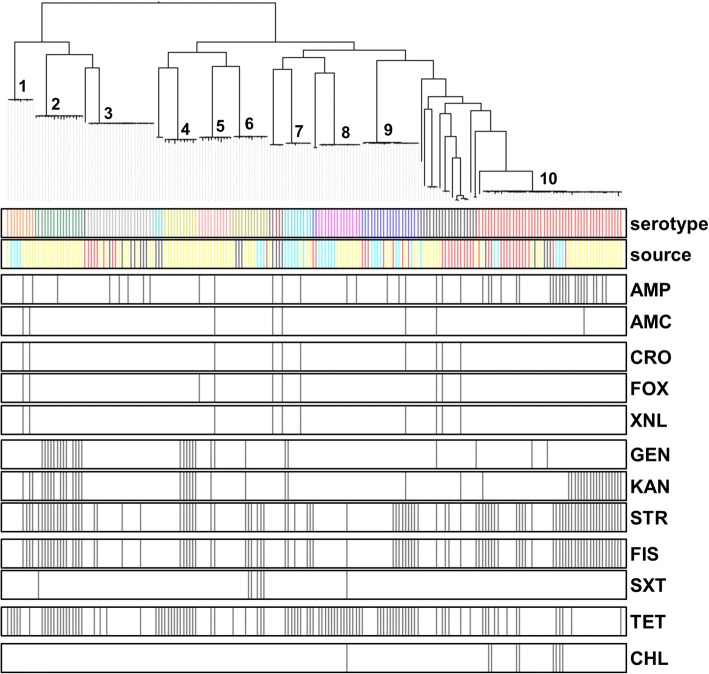
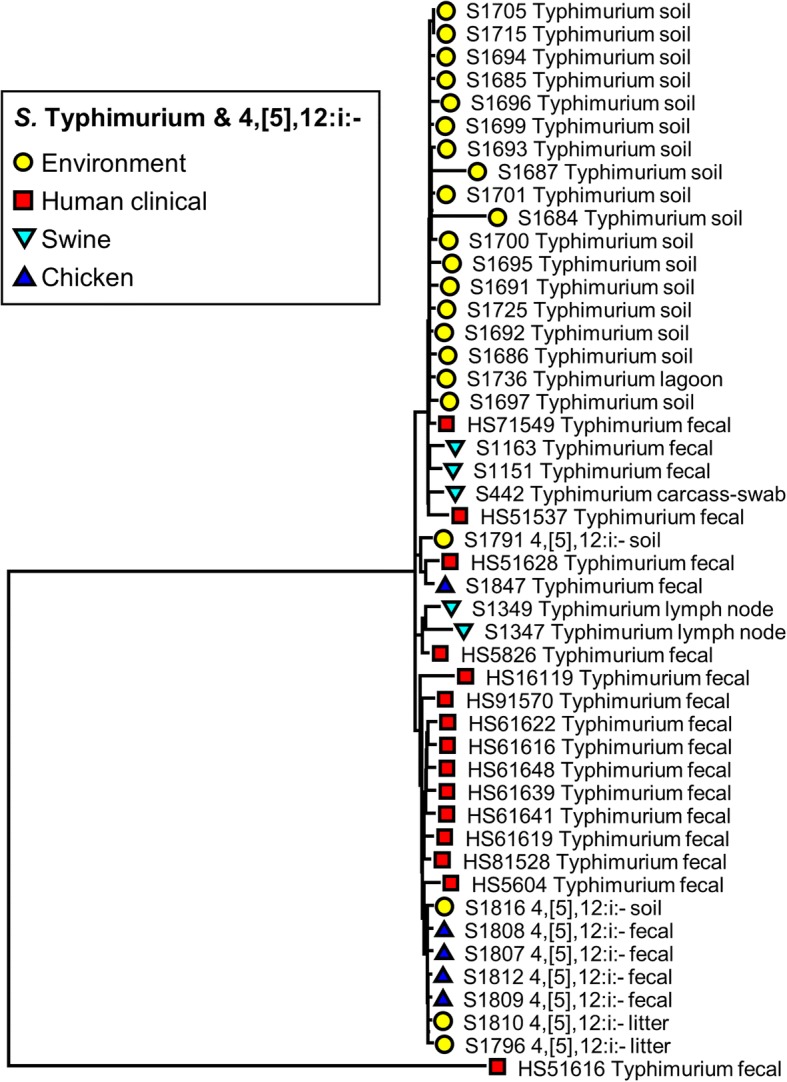
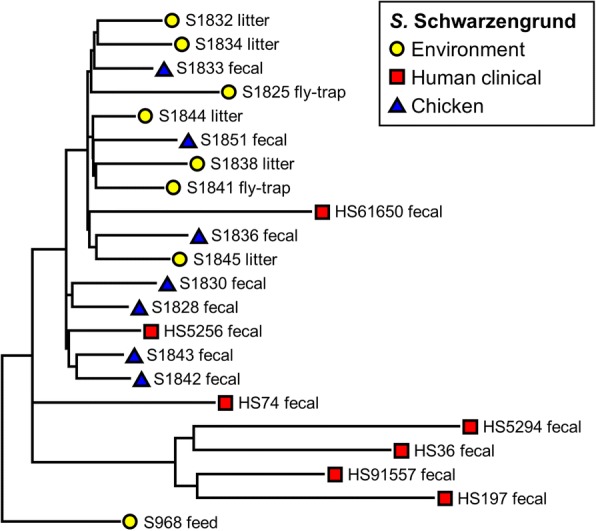
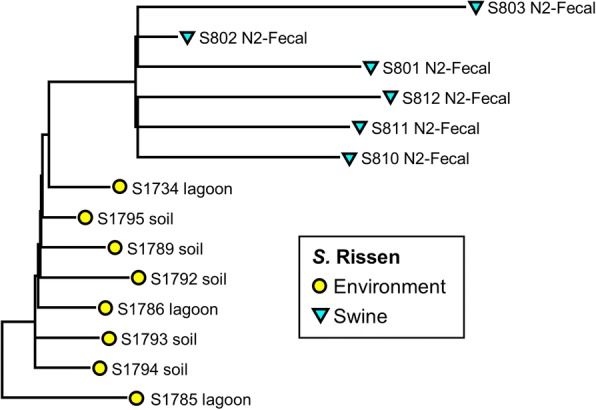
WGS data of 200 S. enterica isolates recovered from human (n = 44), swine (n = 32), poultry (n = 22), and farm environment (n = 102) were used for in silico prediction of serovar, distribution of virulence genes, and phylogenetically clustered using core genome single nucleotide polymorphism (SNP) and feature frequency profiling (FFP). Furthermore, AMR was studied both by genotypic prediction using five curated AMR databases, and compared to phenotypic AMR using broth microdilution. Core genome SNP-based and FFP-based phylogenetic trees showed consistent clustering of isolates into the respective serovars, and suggested clustering of isolates based on the source of isolation. The overall correlation of phenotypic and genotypic AMR was 87.61% and 97.13% for sensitivity and specificity, respectively. AMR and virulence genes clustered with the Salmonella serovars, while there were also associations between the presence of virulence genes in both animal/environmental isolates and human clinical samples.
WGS is a helpful tool for Salmonella phylogenetic analysis, AMR and virulence gene predictions. The clinical isolates clustered closely with animal and environmental isolates, suggesting that animals and environment are potential sources for dissemination of AMR and virulence genes between Salmonella serovars.
沙门氏菌是一种重要的食源性病原体,可通过多种不同途径传播,关于获得抗生素耐药性(AMR)的报告正在增加。为了更好地了解人类沙门氏菌临床分离株与潜在的环境/动物宿主之间的关系,我们使用全基因组测序(WGS)来研究来自美国两个相邻州的沙门氏菌分离株的流行病学和 AMR 模式。
使用来自人类(n=44)、猪(n=32)、家禽(n=22)和农场环境(n=102)的 200 个 S. enterica 分离株的 WGS 数据,对血清型、毒力基因分布进行了计算机预测,并用核心基因组单核苷酸多态性(SNP)和特征频率分析(FFP)进行了系统发育聚类。此外,我们使用五个经过验证的 AMR 数据库进行了基因型预测来研究 AMR,并用肉汤微量稀释法进行了表型 AMR 比较。基于核心基因组 SNP 的和基于 FFP 的系统发育树一致地将分离株聚类到各自的血清型中,并提示根据分离株的来源进行聚类。表型和基因型 AMR 的总体相关性分别为敏感性 87.61%和特异性 97.13%。AMR 和毒力基因与沙门氏菌血清型聚类,同时动物/环境分离株和人类临床样本中也存在毒力基因的存在。
WGS 是沙门氏菌系统发育分析、AMR 和毒力基因预测的有用工具。临床分离株与动物和环境分离株紧密聚类,表明动物和环境是沙门氏菌血清型之间 AMR 和毒力基因传播的潜在来源。