Department of Microbiology and Immunology, McGill University, 3775 University Street, H3A 2B4, Montreal, Quebec, Canada.
Department of Chemistry and Biochemistry, University of Oklahoma, Norman, OK, USA.
Sci Rep. 2018 Nov 8;8(1):16549. doi: 10.1038/s41598-018-34812-x.
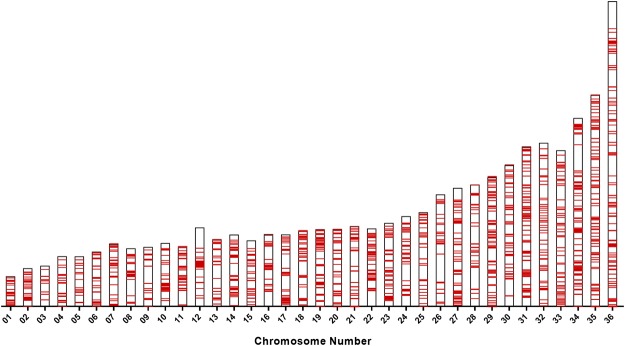
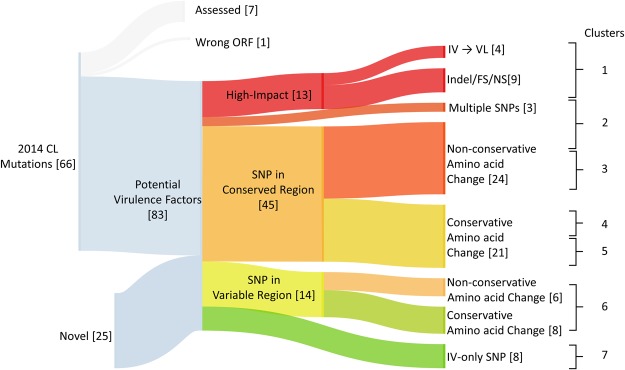
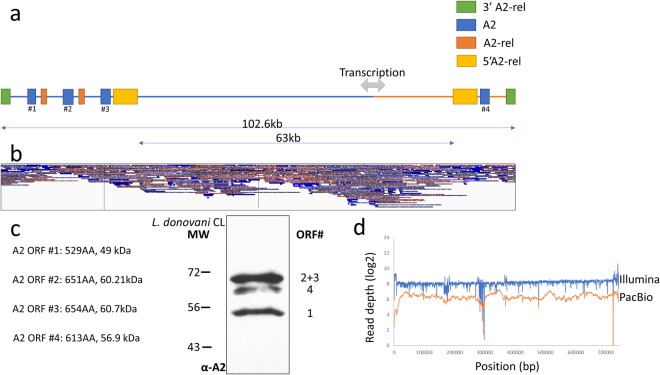
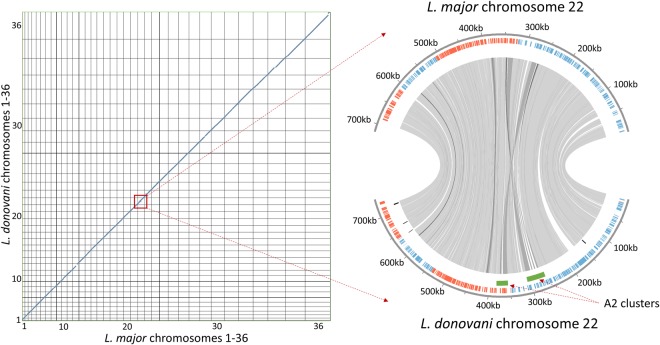
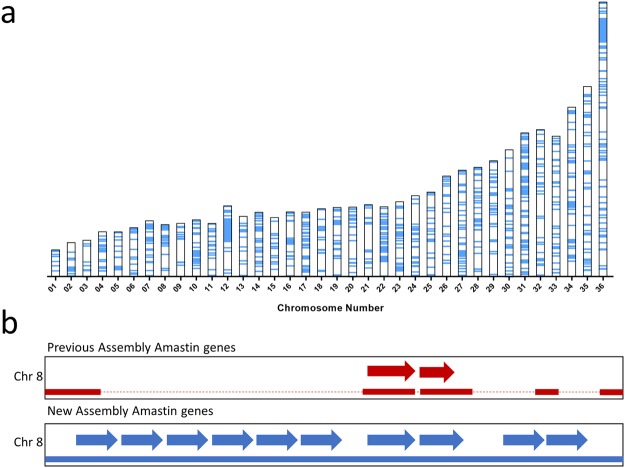
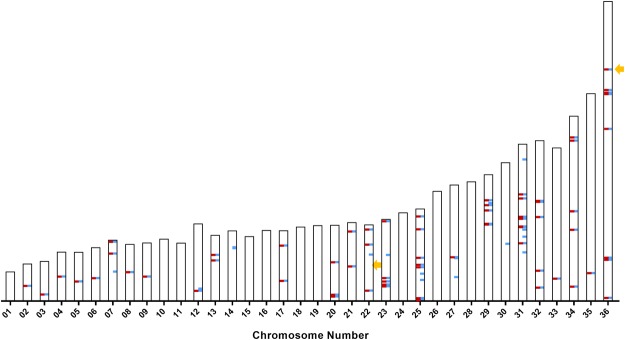
Leishmania donovani is responsible for visceral leishmaniasis, a neglected and lethal parasitic disease with limited treatment options and no vaccine. The study of L. donovani has been hindered by the lack of a high-quality reference genome and this can impact experimental outcomes including the identification of virulence genes, drug targets and vaccine development. We therefore generated a complete genome assembly by deep sequencing using a combination of second generation (Illumina) and third generation (PacBio) sequencing technologies. Compared to the current L. donovani assembly, the genome assembly reported within resulted in the closure over 2,000 gaps, the extension of several chromosomes up to telomeric repeats and the re-annotation of close to 15% of protein coding genes and the annotation of hundreds of non-coding RNA genes. It was possible to correctly assemble the highly repetitive A2 and Amastin virulence gene clusters. A comparative sequence analysis using the improved reference genome confirmed 70 published and identified 15 novel genomic differences between closely related visceral and atypical cutaneous disease-causing L. donovani strains providing a more complete map of genes associated with virulence and visceral organ tropism. Bioinformatic tools including protein variation effect analyzer and basic local alignment search tool were used to prioritize a list of potential virulence genes based on mutation severity, gene conservation and function. This complete genome assembly and novel information on virulence factors will support the identification of new drug targets and the development of a vaccine for L. donovani.
杜氏利什曼原虫是内脏利什曼病的病原体,这是一种被忽视的致命寄生虫病,治疗选择有限,且尚无疫苗。由于缺乏高质量的参考基因组,杜氏利什曼原虫的研究受到了阻碍,这可能会影响实验结果,包括毒力基因、药物靶点和疫苗开发的鉴定。因此,我们使用第二代(Illumina)和第三代(PacBio)测序技术的组合进行深度测序,生成了一个完整的基因组组装。与当前的利什曼原虫组装相比,报告的基因组组装结果封闭了 2000 多个缺口,将几个染色体延伸到端粒重复序列,并重新注释了近 15%的蛋白质编码基因和数百个非编码 RNA 基因。能够正确组装高度重复的 A2 和 Amastin 毒力基因簇。使用改进的参考基因组进行的比较序列分析证实了 70 个已发表的和鉴定了 15 个新的基因组差异,这些差异存在于密切相关的内脏和非典型皮肤疾病引起的利什曼原虫菌株之间,提供了与毒力和内脏器官嗜性相关的基因的更完整图谱。生物信息学工具,包括蛋白质变异效应分析器和基本局部比对搜索工具,用于根据突变严重程度、基因保守性和功能,优先列出潜在毒力基因列表。这个完整的基因组组装和新的毒力因子信息将支持利什曼原虫新药靶的鉴定和疫苗的开发。