School of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai 201418, China.
Molecules. 2018 Nov 9;23(11):2924. doi: 10.3390/molecules23112924.
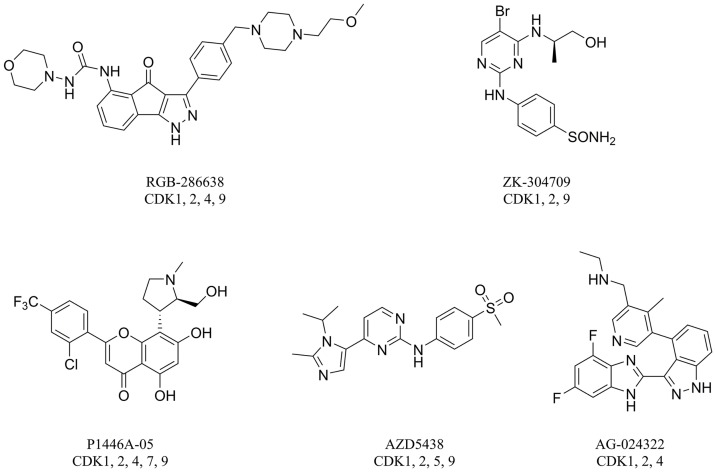
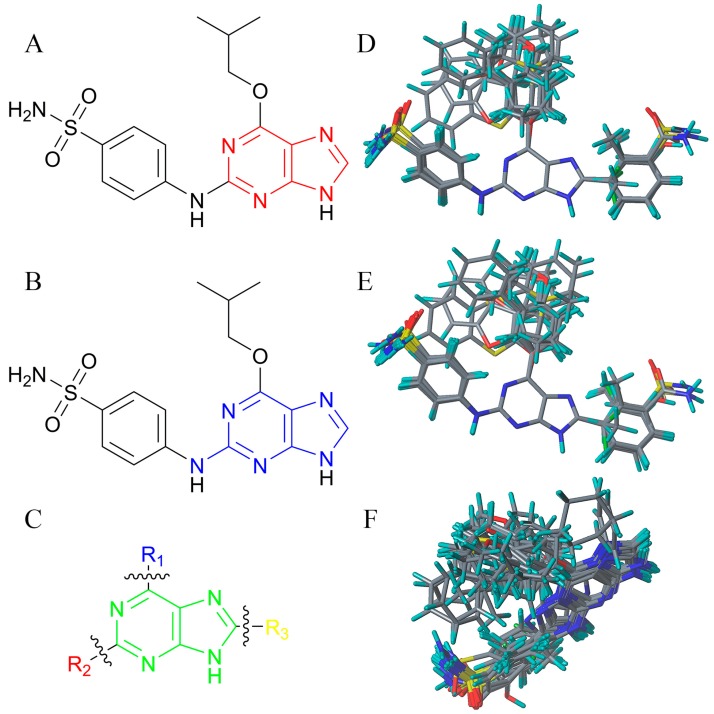
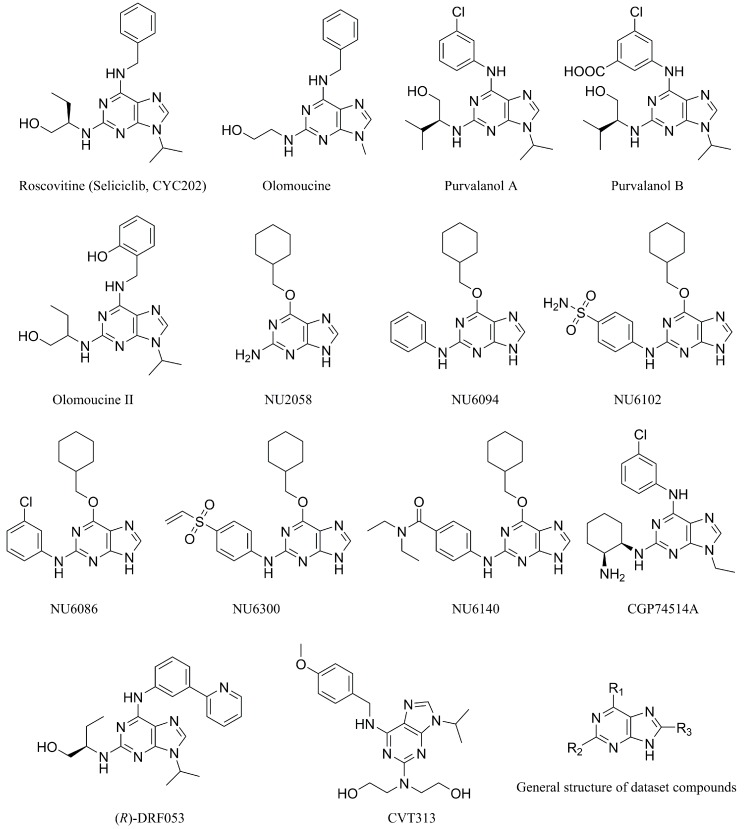
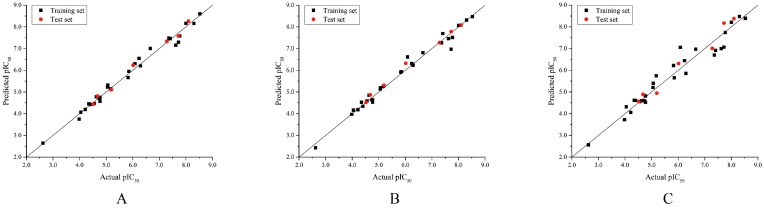
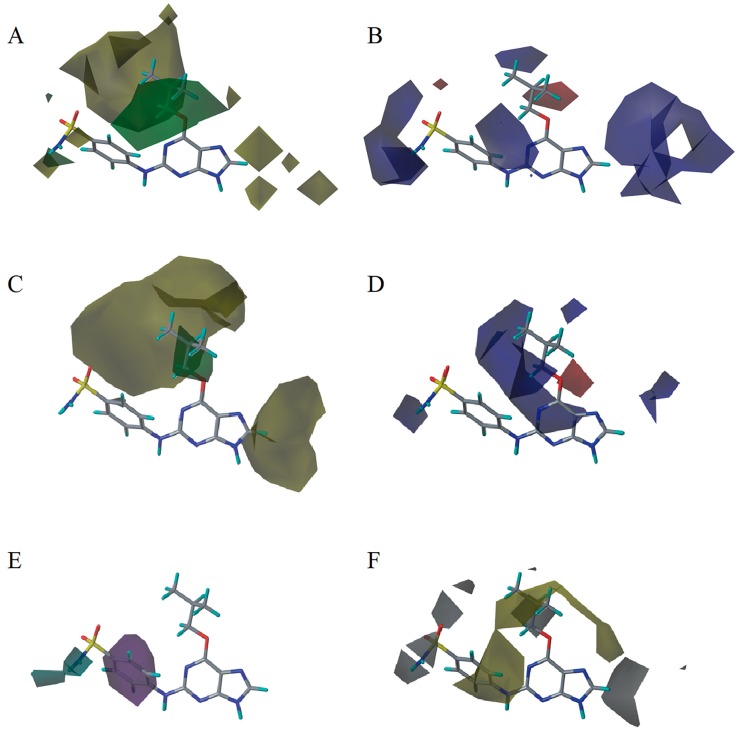
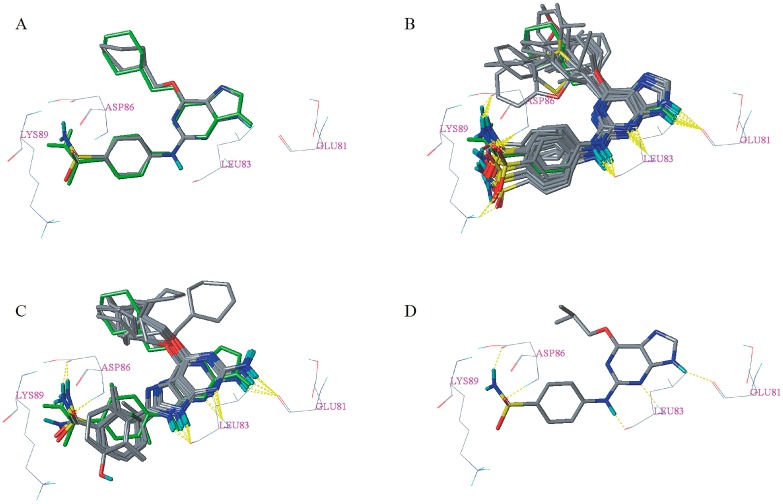
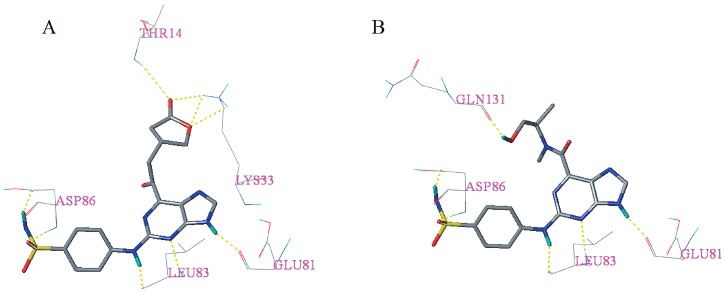
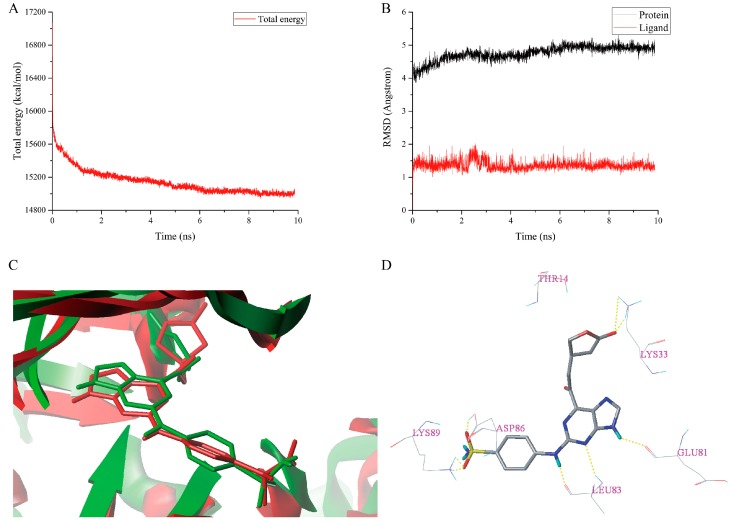
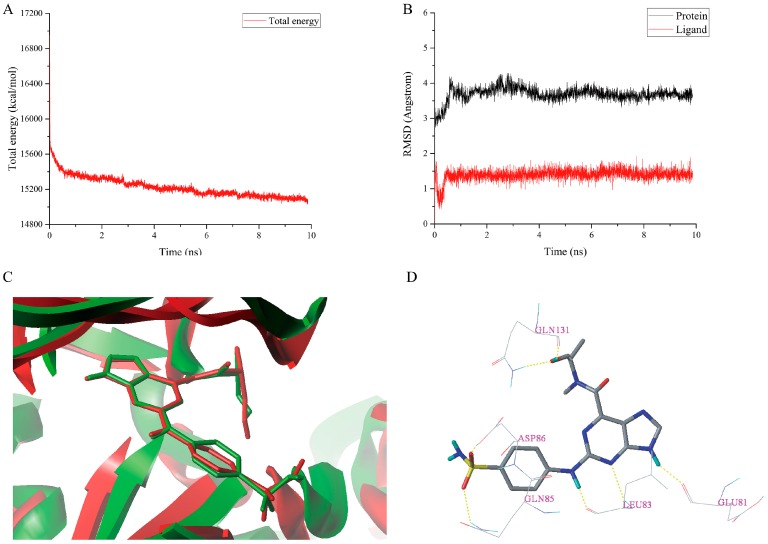
Cyclin-dependent kinase 2 (CDK2) is a potential target for treating cancer. Purine heterocycles have attracted particular attention as the scaffolds for the development of CDK2 inhibitors. To explore the interaction mechanism and the structure⁻activity relationship (SAR) and to design novel candidate compounds as potential CDK2 inhibitors, a systematic molecular modeling study was conducted on 35 purine derivatives as CDK2 inhibitors by combining three-dimensional quantitative SAR (3D-QSAR), virtual screening, molecular docking, and molecular dynamics (MD) simulations. The predictive CoMFA model (q² = 0.743, r pred 2 = 0.991), the CoMSIA model (q² = 0.808, r pred 2 = 0.990), and the Topomer CoMFA model (q² = 0.779, r pred 2 = 0.962) were obtained. Contour maps revealed that the electrostatic, hydrophobic, hydrogen bond donor and steric fields played key roles in the QSAR models. Thirty-one novel candidate compounds with suitable predicted activity (predicted pIC > 8) were designed by using the results of virtual screening. Molecular docking indicated that residues Asp86, Glu81, Leu83, Lys89, Lys33, and Gln131 formed hydrogen bonds with the ligand, which affected activity of the ligand. Based on the QSAR model prediction and molecular docking, two candidate compounds, and (predicted pIC > 8, docking score > 10), with the most potential research value were further screened out. MD simulations of the corresponding complexes of these two candidate compounds further verified their stability. This study provided valuable information for the development of new potential CDK2 inhibitors.
细胞周期蛋白依赖性激酶 2(CDK2)是治疗癌症的潜在靶点。嘌呤杂环因其作为 CDK2 抑制剂开发的骨架而受到特别关注。为了探索相互作用机制和结构-活性关系(SAR),并设计新型候选化合物作为潜在的 CDK2 抑制剂,通过将三维定量构效关系(3D-QSAR)、虚拟筛选、分子对接和分子动力学(MD)模拟相结合,对 35 种作为 CDK2 抑制剂的嘌呤衍生物进行了系统的分子建模研究。预测性 CoMFA 模型(q² = 0.743,r pred 2 = 0.991)、CoMSIA 模型(q² = 0.808,r pred 2 = 0.990)和 Topomer CoMFA 模型(q² = 0.779,r pred 2 = 0.962)被获得。轮廓图显示,静电、疏水、氢键供体和立体场在 QSAR 模型中起着关键作用。通过虚拟筛选的结果,设计了 31 种具有合适预测活性(预测 pIC > 8)的新型候选化合物。分子对接表明,残基 Asp86、Glu81、Leu83、Lys89、Lys33 和 Gln131 与配体形成氢键,影响配体的活性。基于 QSAR 模型预测和分子对接,进一步筛选出两个候选化合物和(预测 pIC > 8,对接得分 > 10),它们具有最大的潜在研究价值。这两种候选化合物相应配合物的 MD 模拟进一步验证了它们的稳定性。本研究为开发新的潜在 CDK2 抑制剂提供了有价值的信息。