Frontier Laboratories for Value Creation, Sapporo Holdings Ltd., Yaizu, Shizuoka, 425-0013, Japan.
Dairy Science and Technology Institute, Kyodo Milk Industry Co. Ltd., Hinode-machi, Tokyo, 190-0182, Japan.
BMC Microbiol. 2018 Nov 16;18(1):188. doi: 10.1186/s12866-018-1311-8.
16S rRNA gene amplicon sequencing analysis (16S amplicon sequencing) has provided considerable information regarding the ecology of the intestinal microbiome. Recently, metabolomics has been used for investigating the crosstalk between the intestinal microbiome and the host via metabolites. In the present study, we determined the accuracy with which 16S rRNA gene data at different classification levels correspond to the metabolome data for an in-depth understanding of the intestinal environment.
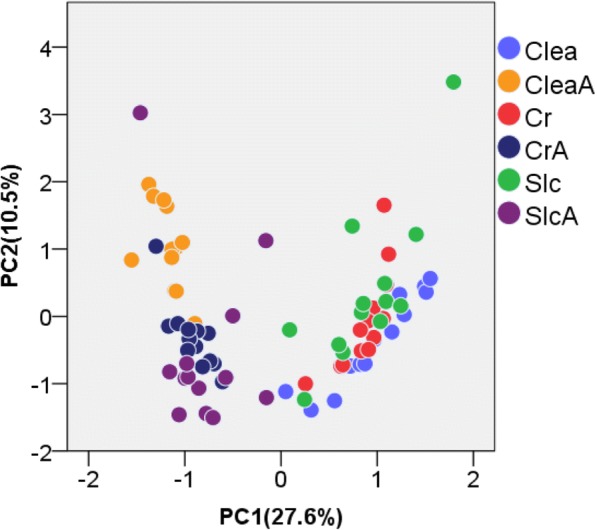
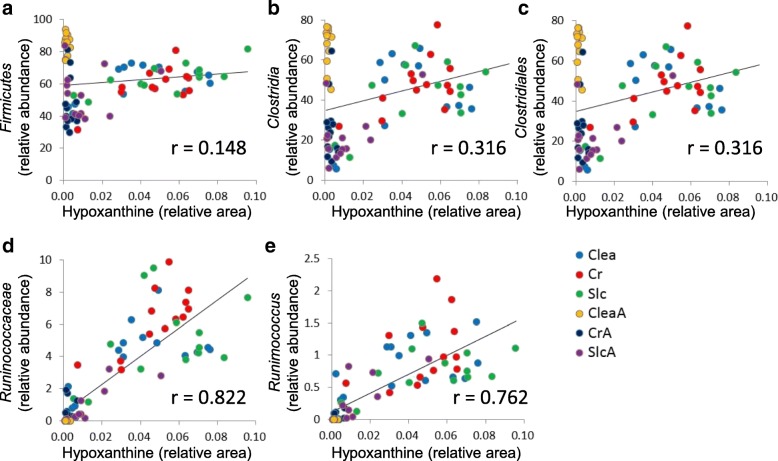
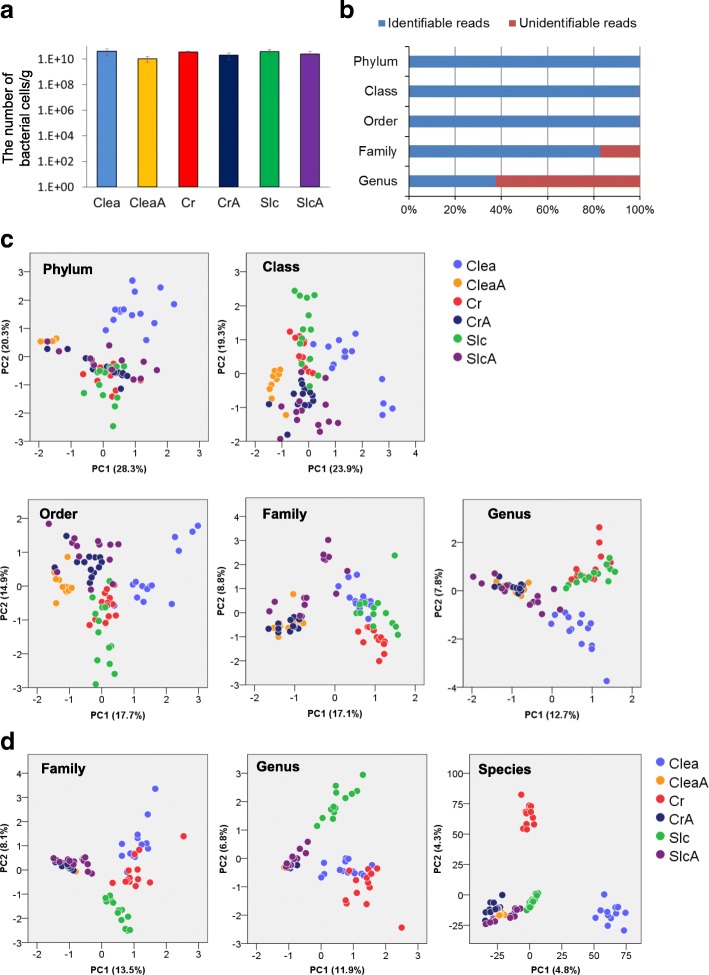
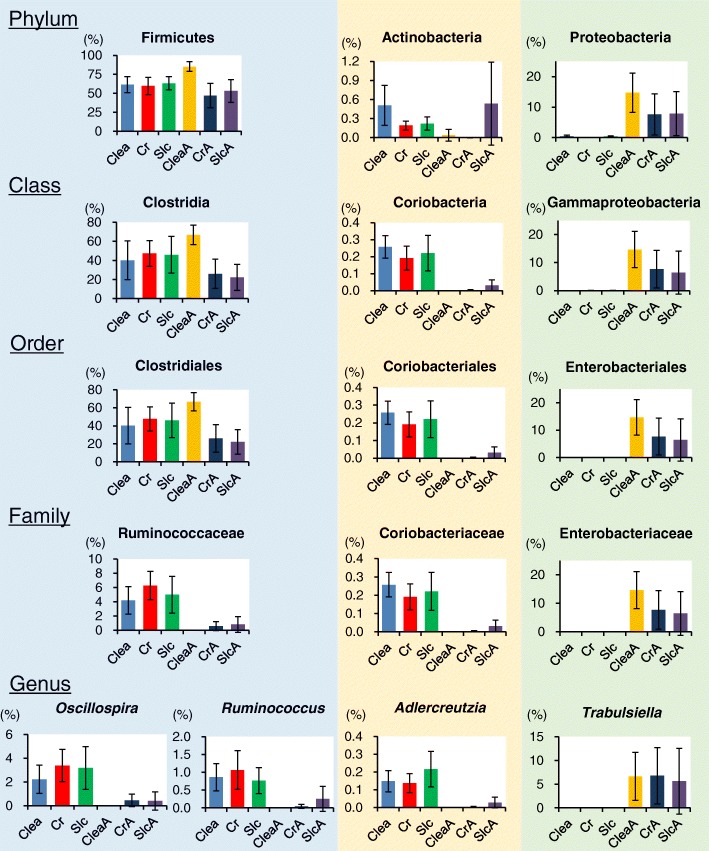
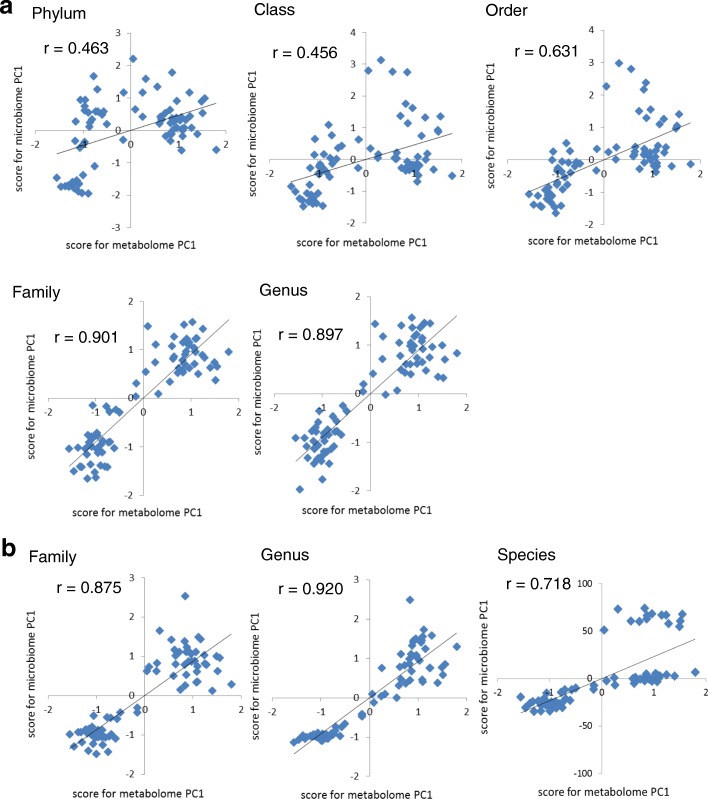
Over 200 metabolites were identified using capillary electrophoresis and time-of-flight mass spectrometry (CE-TOFMS)-based metabolomics in the feces of antibiotic-treated and untreated mice. 16S amplicon sequencing, followed by principal component analysis (PCA) of the intestinal microbiome at each taxonomic rank, revealed differences between the antibiotic-treated and untreated groups in the first principal component in the family-, genus, and species-level analyses. These differences were similar to those observed in the PCA of the metabolome. Furthermore, a strong correlation between principal component (PC) scores of the metabolome and microbiome was observed in family-, genus-, and species-level analyses.
Lower taxonomic ranks such as family, genus, or species are preferable for 16S amplicon sequencing to investigate the correlation between the microbiome and metabolome. The correlation of PC scores between the microbiome and metabolome at lower taxonomic levels yield a simple method of integrating different "-omics" data, which provides insights regarding crosstalk between the intestinal microbiome and the host.
16S rRNA 基因扩增子测序分析(16S 扩增子测序)为肠道微生物组的生态学提供了大量信息。最近,代谢组学已被用于通过代谢物研究肠道微生物组与宿主之间的串扰。在本研究中,我们确定了不同分类水平的 16S rRNA 基因数据与代谢组数据的对应程度的准确性,以深入了解肠道环境。
采用毛细管电泳-飞行时间质谱(CE-TOFMS)代谢组学方法在抗生素处理和未处理的小鼠粪便中鉴定出超过 200 种代谢物。16S 扩增子测序,然后对每个分类等级的肠道微生物组进行主成分分析(PCA),在科、属和种水平的分析中,在第一主成分上显示出抗生素处理组和未处理组之间的差异。这些差异与代谢组 PCA 中观察到的相似。此外,在科、属和种水平的分析中,代谢组和微生物组的主成分(PC)得分之间存在很强的相关性。
较低的分类等级,如科、属或种,更适合 16S 扩增子测序来研究微生物组和代谢组之间的相关性。在较低的分类水平上,微生物组和代谢组的 PC 得分之间的相关性提供了一种简单的方法来整合不同的“组学”数据,从而深入了解肠道微生物组与宿主之间的串扰。