Wang Zeya, Cao Shaolong, Morris Jeffrey S, Ahn Jaeil, Liu Rongjie, Tyekucheva Svitlana, Gao Fan, Li Bo, Lu Wei, Tang Ximing, Wistuba Ignacio I, Bowden Michaela, Mucci Lorelei, Loda Massimo, Parmigiani Giovanni, Holmes Chris C, Wang Wenyi
Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA; Department of Statistics, Rice University, Houston, TX 77005, USA.
Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
iScience. 2018 Nov 30;9:451-460. doi: 10.1016/j.isci.2018.10.028. Epub 2018 Nov 2.
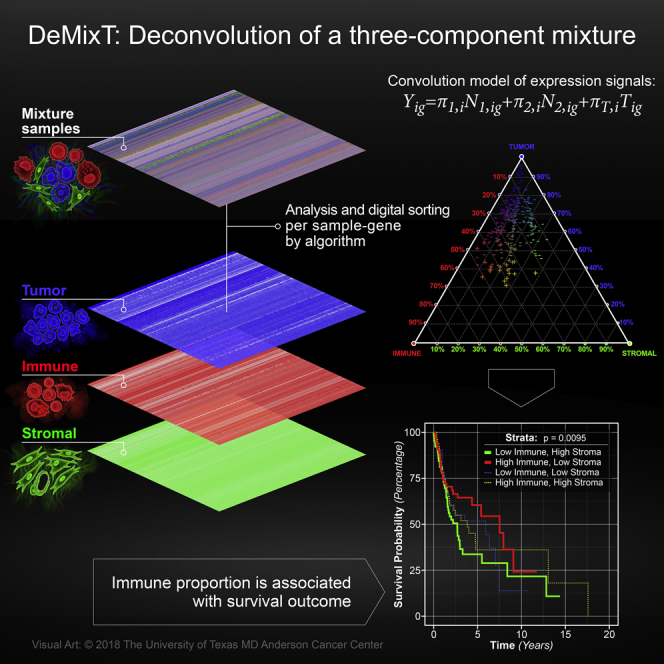
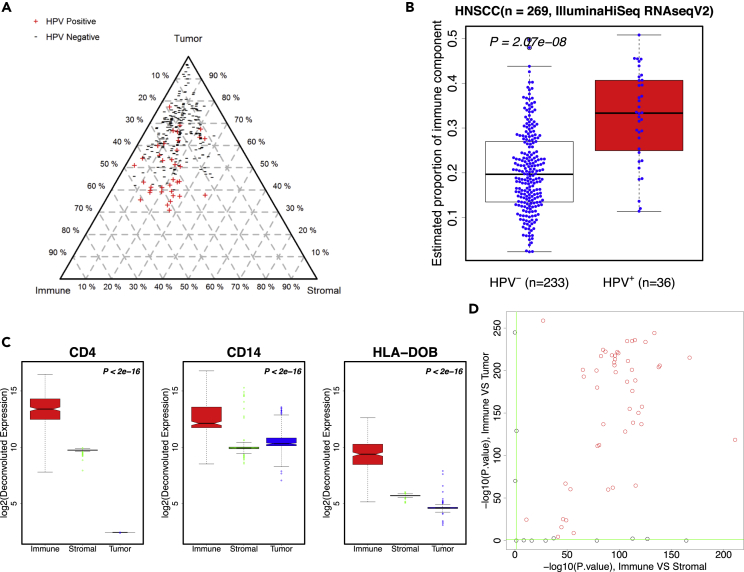
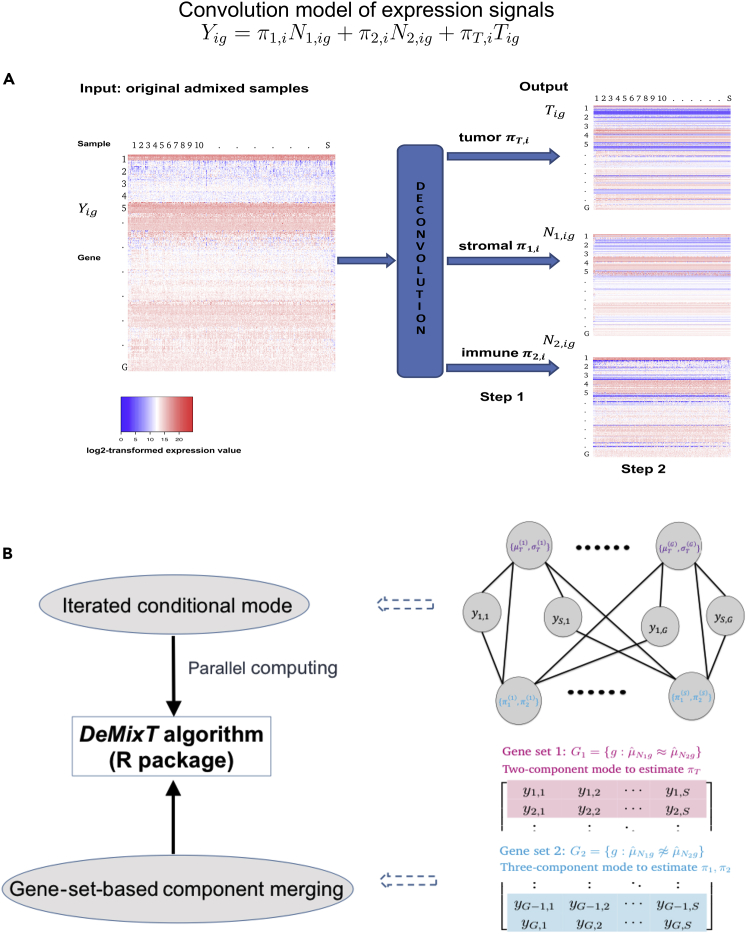
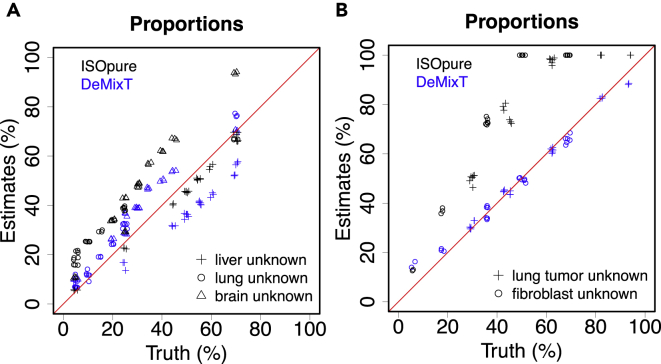
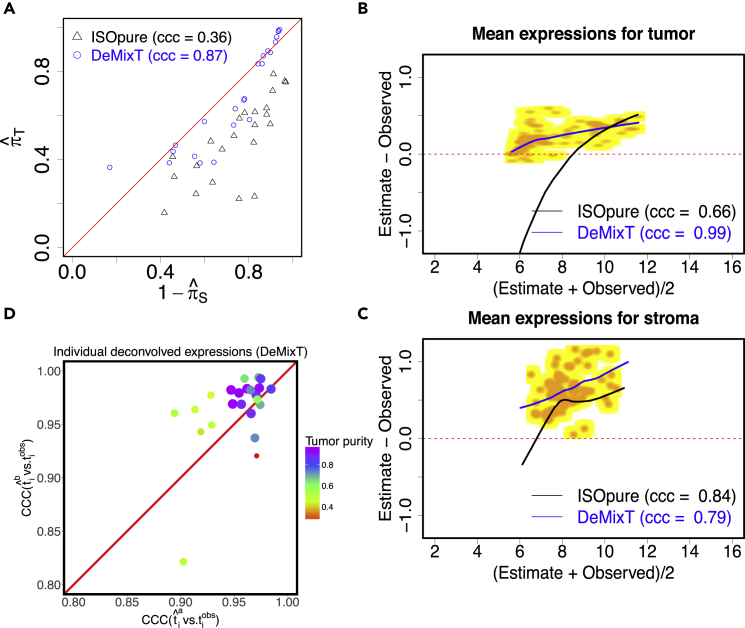
Transcriptome deconvolution in cancer and other heterogeneous tissues remains challenging. Available methods lack the ability to estimate both component-specific proportions and expression profiles for individual samples. We present DeMixT, a new tool to deconvolve high-dimensional data from mixtures of more than two components. DeMixT implements an iterated conditional mode algorithm and a novel gene-set-based component merging approach to improve accuracy. In a series of experimental validation studies and application to TCGA data, DeMixT showed high accuracy. Improved deconvolution is an important step toward linking tumor transcriptomic data with clinical outcomes. An R package, scripts, and data are available: https://github.com/wwylab/DeMixTallmaterials.
癌症及其他异质性组织中的转录组反卷积仍然具有挑战性。现有方法缺乏估计单个样本中各组分特异性比例和表达谱的能力。我们提出了DeMixT,这是一种用于对来自两种以上组分混合物的高维数据进行反卷积的新工具。DeMixT实现了一种迭代条件模式算法和一种基于基因集的新型组分合并方法以提高准确性。在一系列实验验证研究以及对TCGA数据的应用中,DeMixT显示出了高准确性。改进的反卷积是将肿瘤转录组数据与临床结果相联系的重要一步。可获取一个R包、脚本和数据:https://github.com/wwylab/DeMixTallmaterials 。