Said Mohammed Khadija, Kibinge Nelson, Prins Pjotr, Agoti Charles N, Cotten Matthew, Nokes D J, Brand Samuel, Githinji George
Pwani University, Kilifi, Kenya.
KEMRI-Wellcome Trust Research Programme, KEMRI Centre for Geographic Medicine Research - Coast, Kilifi, Kenya.
Wellcome Open Res. 2018 Sep 13;3:21. doi: 10.12688/wellcomeopenres.13538.2. eCollection 2018.
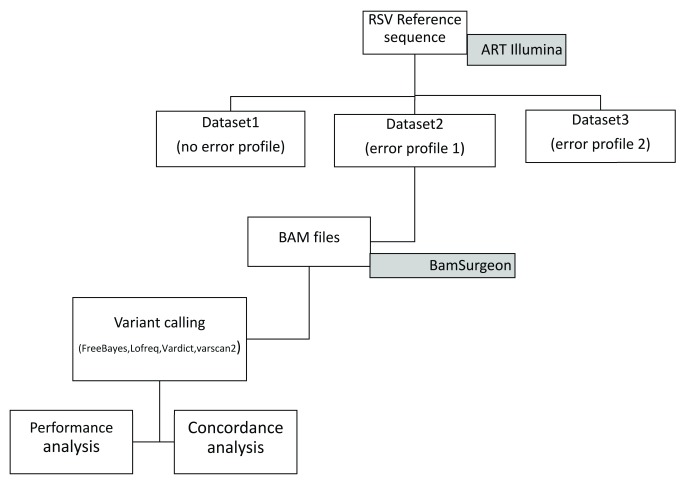
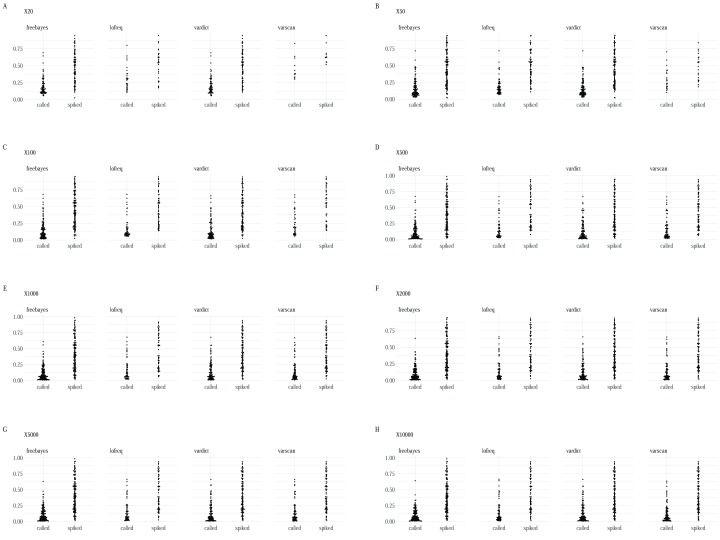
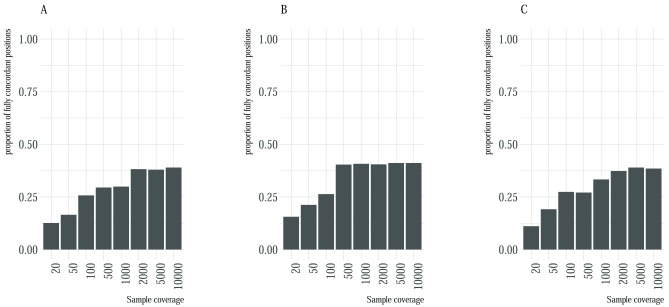
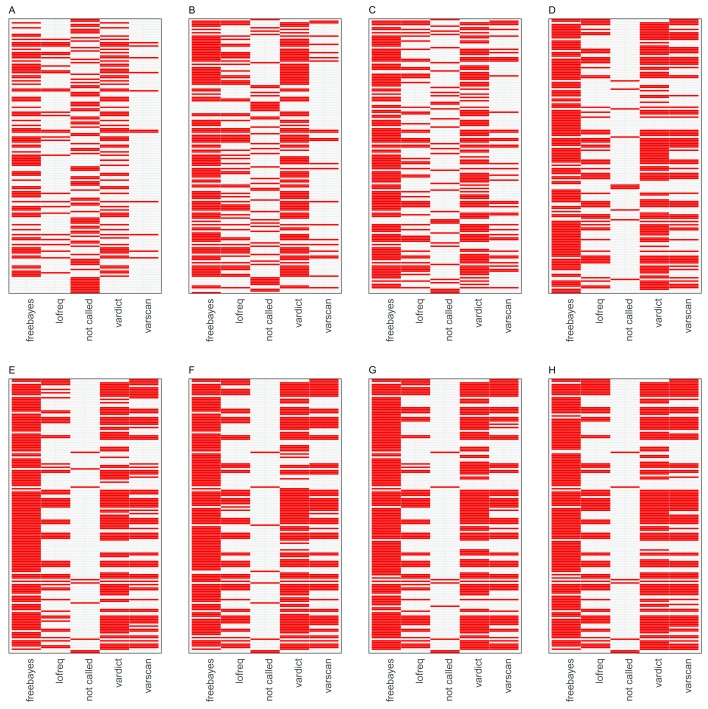
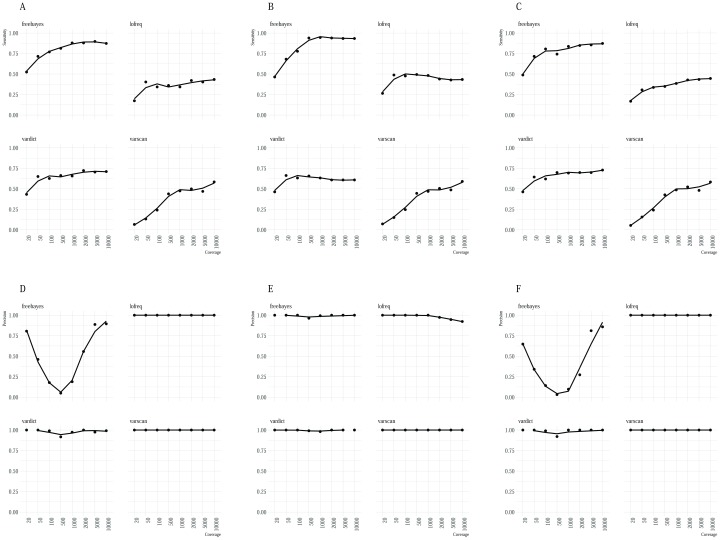
High-throughput whole genome sequencing facilitates investigation of minority virus sub-populations from virus positive samples. Minority variants are useful in understanding within and between host diversity, population dynamics and can potentially assist in elucidating person-person transmission pathways. Several minority variant callers have been developed to describe low frequency sub-populations from whole genome sequence data. These callers differ based on bioinformatics and statistical methods used to discriminate sequencing errors from low-frequency variants. We evaluated the diagnostic performance and concordance between published minority variant callers used in identifying minority variants from whole-genome sequence data from virus samples. We used the ART-Illumina read simulation tool to generate three artificial short-read datasets of varying coverage and error profiles from an RSV reference genome. The datasets were spiked with nucleotide variants at predetermined positions and frequencies. Variants were called using FreeBayes, LoFreq, Vardict, and VarScan2. The variant callers' agreement in identifying known variants was quantified using two measures; concordance accuracy and the inter-caller concordance. The variant callers reported differences in identifying minority variants from the datasets. Concordance accuracy and inter-caller concordance were positively correlated with sample coverage. FreeBayes identified the majority of variants although it was characterised by variable sensitivity and precision in addition to a high false positive rate relative to the other minority variant callers and which varied with sample coverage. LoFreq was the most conservative caller. We conducted a performance and concordance evaluation of four minority variant calling tools used to identify and quantify low frequency variants. Inconsistency in the quality of sequenced samples impacts on sensitivity and accuracy of minority variant callers. Our study suggests that combining at least three tools when identifying minority variants is useful in filtering errors when calling low frequency variants.
高通量全基因组测序有助于对病毒阳性样本中的少数病毒亚群进行研究。少数变异体有助于理解宿主内部和宿主之间的多样性、群体动态,并且可能有助于阐明人际传播途径。已经开发了几种少数变异体检测工具来描述全基因组序列数据中的低频亚群。这些检测工具因用于区分测序错误和低频变异体的生物信息学和统计方法而异。我们评估了已发表的少数变异体检测工具在从病毒样本的全基因组序列数据中识别少数变异体时的诊断性能和一致性。我们使用ART-Illumina读段模拟工具从呼吸道合胞病毒(RSV)参考基因组生成了三个具有不同覆盖度和错误谱的人工短读段数据集。这些数据集在预定位置和频率处掺入了核苷酸变异体。使用FreeBayes、LoFreq、Vardict和VarScan2对变异体进行检测。使用两种方法对变异体检测工具在识别已知变异体方面的一致性进行了量化;一致性准确性和检测工具间的一致性。变异体检测工具在从数据集中识别少数变异体方面存在差异。一致性准确性和检测工具间的一致性与样本覆盖度呈正相关。FreeBayes识别出了大多数变异体,尽管其具有可变的灵敏度和精确性,并且相对于其他少数变异体检测工具具有较高的假阳性率,且该假阳性率随样本覆盖度而变化。LoFreq是最保守的检测工具。我们对用于识别和量化低频变异体的四种少数变异体检测工具进行了性能和一致性评估。测序样本质量的不一致会影响少数变异体检测工具的灵敏度和准确性。我们的研究表明,在识别少数变异体时至少结合使用三种工具,有助于在检测低频变异体时过滤错误。