Département de microbiologie-infectiologie et immunologie, Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Québec City, Quebec, Canada.
Centre de Recherche de l'Institut Universitaire de Cardiologie et de Pneumologie de Québec (CRIUCPQ), Québec City, Quebec, Canada.
Genome Biol Evol. 2019 Jan 1;11(1):109-120. doi: 10.1093/gbe/evy259.
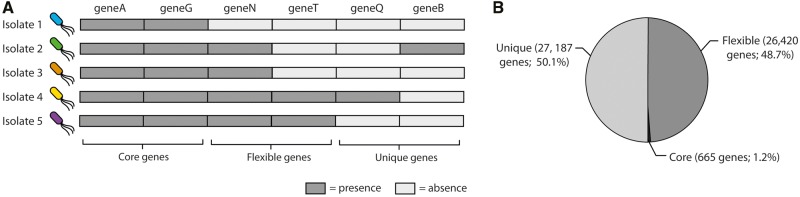
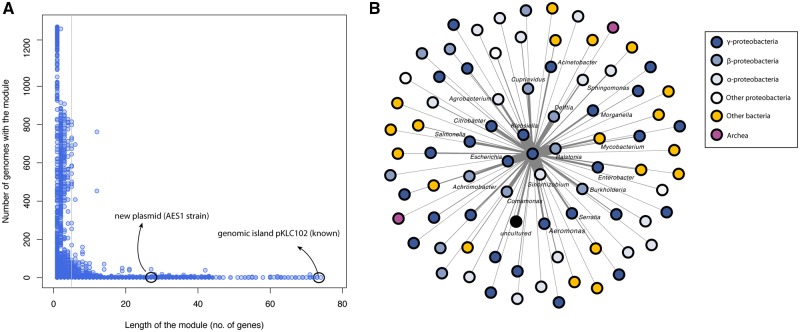
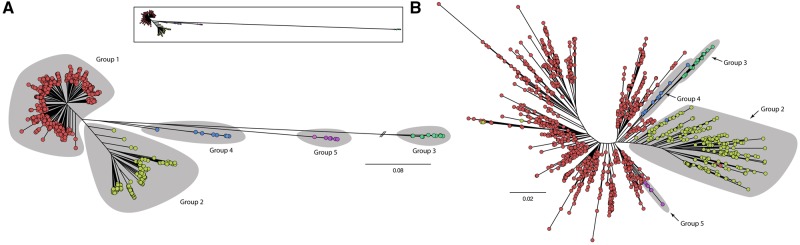
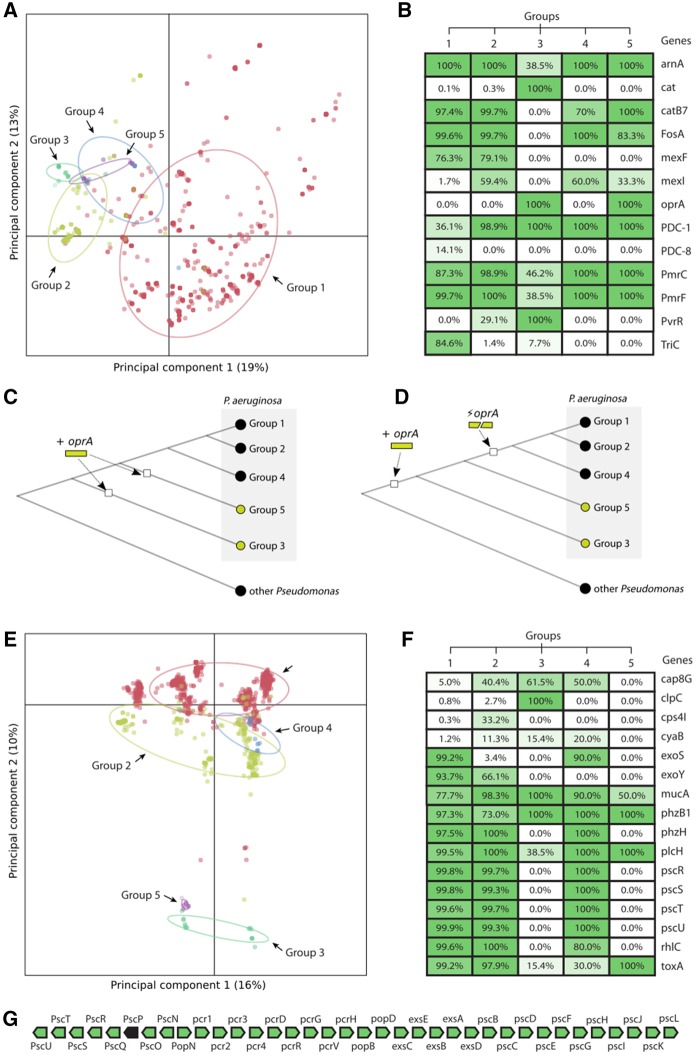
The huge increase in the availability of bacterial genomes led us to a point in which we can investigate and query pan-genomes, for example, the full set of genes of a given bacterial species or clade. Here, we used a data set of 1,311 high-quality genomes from the human pathogen Pseudomonas aeruginosa, 619 of which were newly sequenced, to show that a pan-genomic approach can greatly refine the population structure of bacterial species, provide new insights to define species boundaries, and generate hypotheses on the evolution of pathogenicity. The 665-gene P. aeruginosa core genome presented here, which constitutes only 1% of the entire pan-genome, is the first to be in the same order of magnitude as the minimal bacterial genome and represents a conservative estimate of the actual core genome. Moreover, the phylogeny based on this core genome provides strong evidence for a five-group population structure that includes two previously undescribed groups of isolates. Comparative genomics focusing on antimicrobial resistance and virulence genes showed that variation among isolates was partly linked to this population structure. Finally, we hypothesized that horizontal gene transfer had an important role in this respect, and found a total of 3,010 putative complete and fragmented plasmids, 5% and 12% of which contained resistance or virulence genes, respectively. This work provides data and strategies to study the evolutionary trajectories of resistance and virulence in P. aeruginosa.
细菌基因组的可用性大量增加,使我们能够研究和查询泛基因组,例如,给定细菌物种或进化枝的全套基因。在这里,我们使用了来自人类病原体铜绿假单胞菌的 1311 个高质量基因组数据集,其中 619 个是新测序的,结果表明泛基因组方法可以大大细化细菌物种的种群结构,为定义物种界限提供新的见解,并对致病性的进化产生假设。这里呈现的 665 个基因的铜绿假单胞菌核心基因组仅占整个泛基因组的 1%,是第一个与最小细菌基因组处于同一数量级的核心基因组,代表了实际核心基因组的保守估计。此外,基于该核心基因组的系统发育提供了强有力的证据,证明存在包括两个以前未描述的分离株群的五组种群结构。关注抗生素耐药性和毒力基因的比较基因组学表明,分离株之间的变异部分与这种种群结构有关。最后,我们假设水平基因转移在这方面发挥了重要作用,总共发现了 3010 个推定的完整和片段质粒,其中分别有 5%和 12%的质粒包含耐药性或毒力基因。这项工作提供了数据和策略来研究铜绿假单胞菌中耐药性和毒力的进化轨迹。