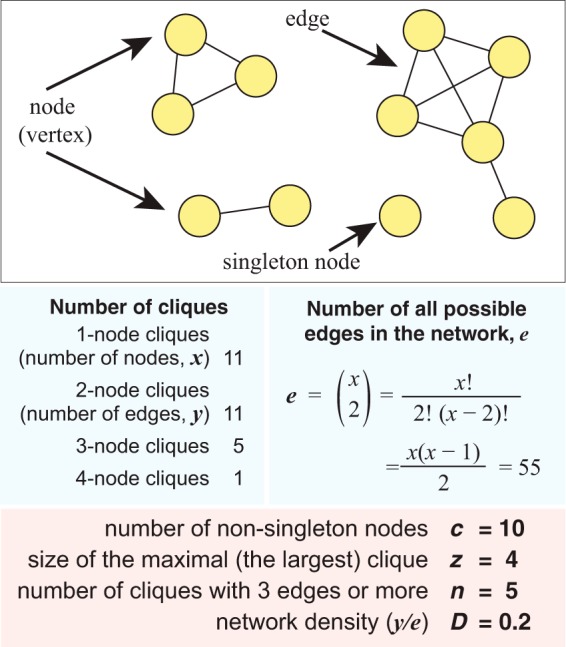
Bernard Guillaume, Greenfield Paul, Ragan Mark A, Chan Cheong Xin
Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Commonwealth Scientific and Industrial Research Organisation (CSIRO), North Ryde, NSW, Australia.
mSystems. 2018 Nov 20;3(6). doi: 10.1128/mSystems.00257-18. eCollection 2018 Nov-Dec.
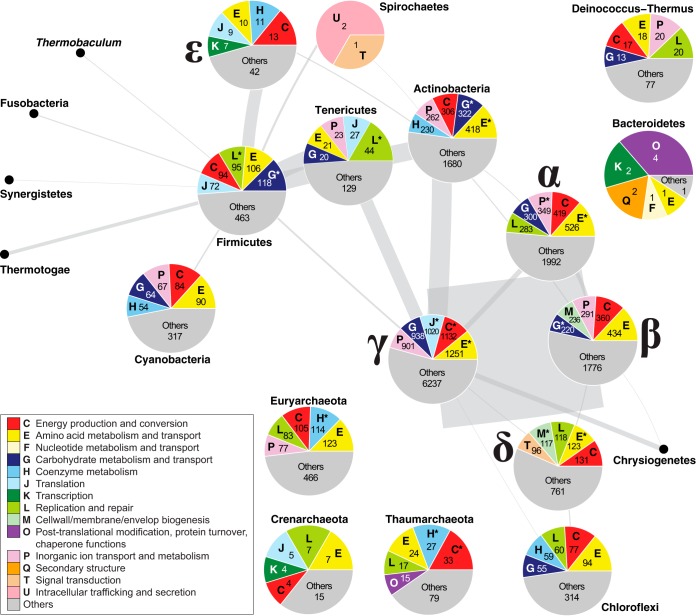
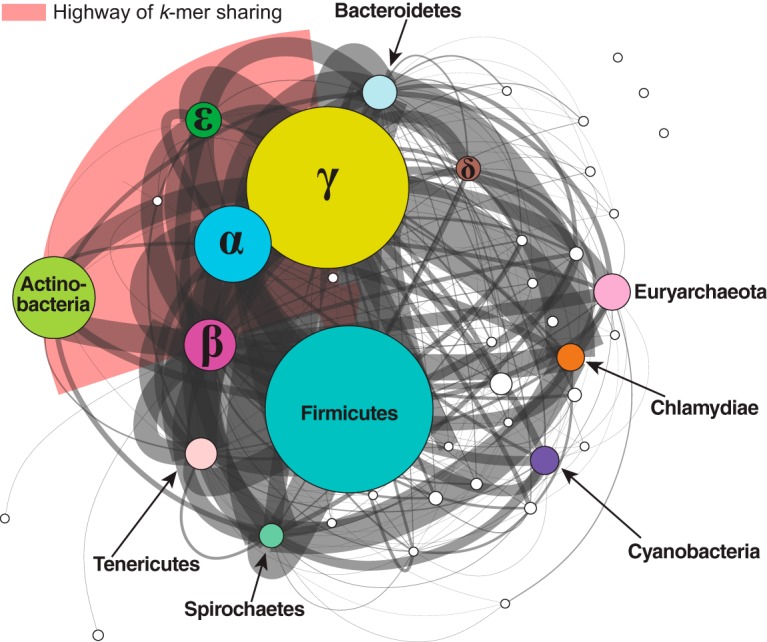
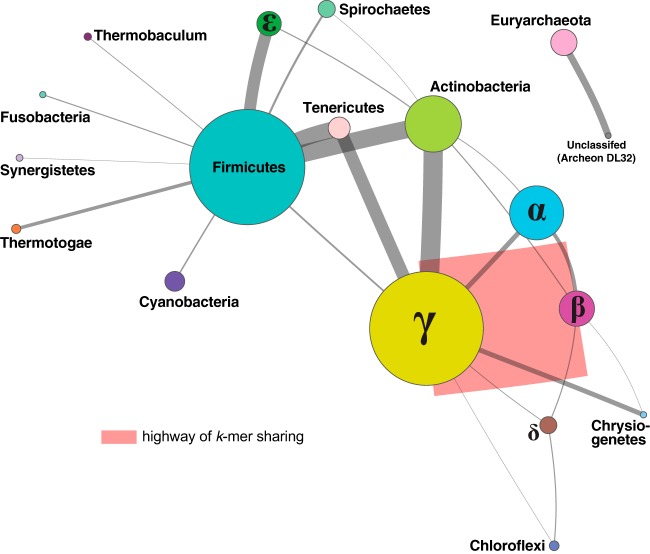
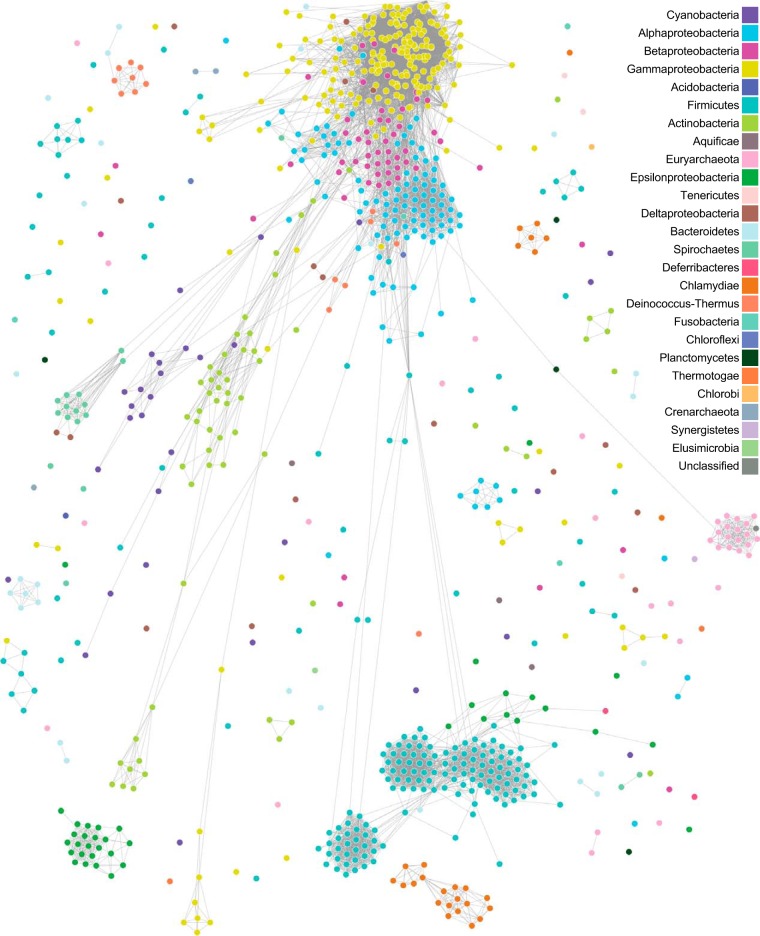
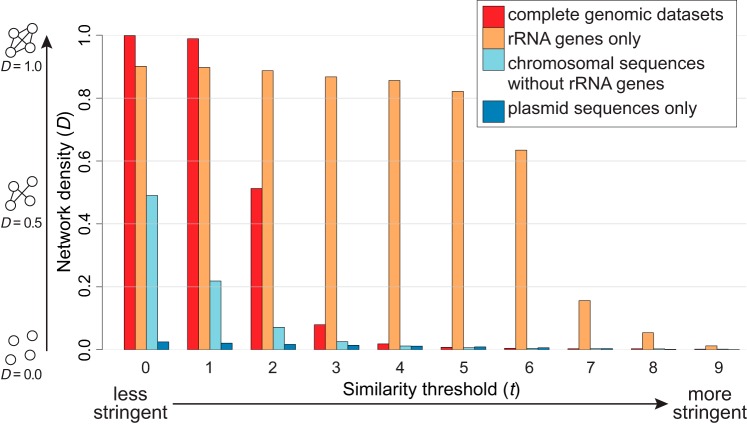
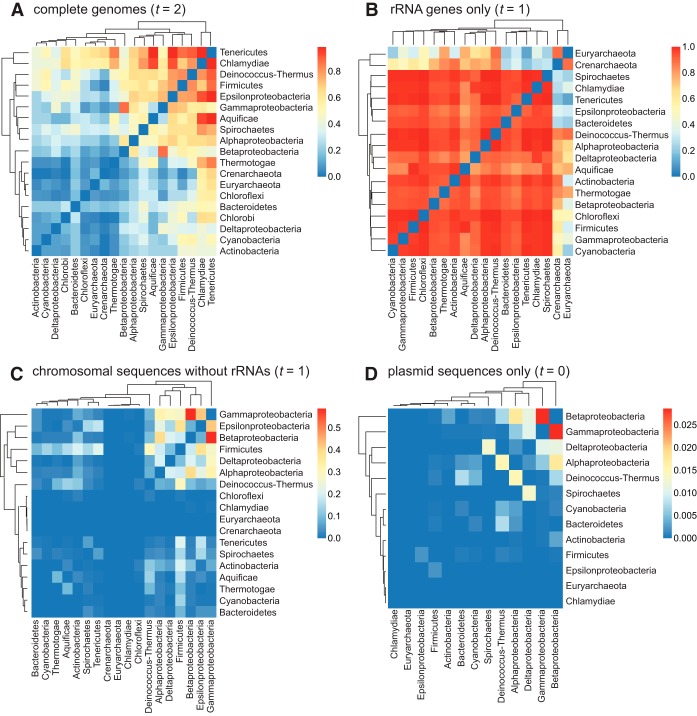
Microbial genomes have been shaped by parent-to-offspring (vertical) descent and lateral genetic transfer. These processes can be distinguished by alignment-based inference and comparison of phylogenetic trees for individual gene families, but this approach is not scalable to whole-genome sequences, and a tree-like structure does not adequately capture how these processes impact microbial physiology. Here we adopted alignment-free approaches based on -mer statistics to infer phylogenomic networks involving 2,783 completely sequenced bacterial and archaeal genomes and compared the contributions of rRNA, protein-coding, and plasmid sequences to these networks. Our results show that the phylogenomic signal arising from ribosomal RNAs is strong and extends broadly across all taxa, whereas that from plasmids is strong but restricted to closely related groups, particularly . However, the signal from the other chromosomal regions is restricted in breadth. We show that mean -mer similarity can correlate with taxonomic rank. We also link the implicated -mers to genome annotation (thus, functions) and define core -mers (thus, core functions) in specific phyletic groups. Highly conserved functions in most phyla include amino acid metabolism and transport as well as energy production and conversion. Intracellular trafficking and secretion are the most prominent core functions among , whereas energy production and conversion are not highly conserved among the largely parasitic or commensal . These observations suggest that differential conservation of functions relates to niche specialization and evolutionary diversification of microbes. Our results demonstrate that -mer approaches can be used to efficiently identify phylogenomic signals and conserved core functions at the multigenome scale. Genome evolution of microbes involves parent-to-offspring descent, and lateral genetic transfer that convolutes the phylogenomic signal. This study investigated phylogenomic signals among thousands of microbial genomes based on short subsequences without using multiple-sequence alignment. The signal from ribosomal RNAs is strong across all taxa, and the signal of plasmids is strong only in closely related groups, particularly . However, the signal from other chromosomal regions (∼99% of the genomes) is remarkably restricted in breadth. The similarity of subsequences is found to correlate with taxonomic rank and informs on conserved and differential core functions relative to niche specialization and evolutionary diversification of microbes. These results provide a comprehensive, alignment-free view of microbial genome evolution as a network, beyond a tree-like structure.
微生物基因组是由亲代到子代(垂直)遗传和横向基因转移塑造而成的。这些过程可以通过基于比对的推断以及对单个基因家族的系统发育树进行比较来区分,但这种方法无法扩展到全基因组序列,而且树状结构也不能充分体现这些过程如何影响微生物生理学。在此,我们采用基于k-mer统计的无比对方法来推断涉及2783个已完全测序的细菌和古菌基因组的系统发育网络,并比较了核糖体RNA、蛋白质编码序列和质粒序列对这些网络的贡献。我们的结果表明,核糖体RNA产生的系统发育信号很强,且广泛延伸至所有分类群,而质粒产生的信号虽强,但仅限于密切相关的群体,尤其是[此处原文缺失相关内容]。然而,来自其他染色体区域的信号在广度上受到限制。我们发现平均k-mer相似性与分类等级相关。我们还将涉及的k-mer与基因组注释(进而与功能)联系起来,并在特定系统发育类群中定义核心k-mer(进而定义核心功能)。大多数门类中高度保守的功能包括氨基酸代谢与转运以及能量产生与转换。细胞内运输和分泌是[此处原文缺失相关内容]中最突出的核心功能,而能量产生与转换在主要为寄生或共生的[此处原文缺失相关内容]中并非高度保守。这些观察结果表明,功能的差异保守性与微生物的生态位特化和进化多样化有关。我们的结果表明,k-mer方法可用于在多基因组规模上有效识别系统发育信号和保守的核心功能。微生物的基因组进化涉及亲代到子代的遗传以及使系统发育信号变得复杂的横向基因转移。本研究基于短序列,在不使用多序列比对的情况下,研究了数千个微生物基因组中的系统发育信号。核糖体RNA产生的信号在所有分类群中都很强,而质粒的信号仅在密切相关的群体中很强,尤其是[此处原文缺失相关内容]。然而,来自其他染色体区域(约占基因组的99%)的信号在广度上受到显著限制。发现子序列的相似性与分类等级相关,并能揭示与微生物的生态位特化和进化多样化相关的保守和差异核心功能。这些结果提供了一个全面的、无比对的微生物基因组进化网络视图,超越了树状结构。