Chung Matthew, Munro James B, Tettelin Hervé, Dunning Hotopp Julie C
Institute for Genome Sciences, University of Maryland Baltimore, Baltimore, Maryland, USA.
Department of Microbiology and Immunology, University of Maryland Baltimore, Baltimore, Maryland, USA.
mSystems. 2018 Dec 4;3(6). doi: 10.1128/mSystems.00236-18. eCollection 2018 Nov-Dec.
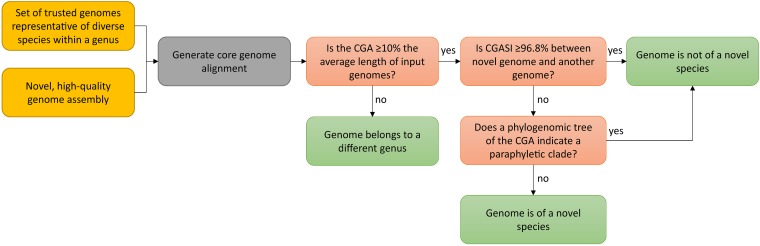
With the exponential increase in the number of bacterial taxa with genome sequence data, a new standardized method to assign species designations is needed that is consistent with classically obtained taxonomic analyses. This is particularly acute for unculturable, obligate intracellular bacteria with which classically defined methods, like DNA-DNA hybridization, cannot be used, such as those in the . In this study, we generated nucleotide-based core genome alignments for a wide range of genera with classically defined species, as well as those within the . We created a workflow that uses the length, sequence identity, and phylogenetic relationships inferred from core genome alignments to assign genus and species designations that recapitulate classically obtained results. Using this method, most classically defined bacterial genera have a core genome alignment that is ≥10% of the average input genome length. Both and fail to meet this criterion, indicating that the taxonomy of these genera should be reexamined. Consistently, genomes from organisms with the same species epithet have ≥96.8% identity of their core genome alignments. Additionally, these core genome alignments can be used to generate phylogenomic trees to identify monophyletic clades that define species and neighbor-network trees to assess recombination across different taxa. By these criteria, organisms are delineated into species different from the currently used supergroup designations, while organisms are delineated into 9 distinct species, compared to the current 27 species. By using core genome alignments to assign taxonomic designations, we aim to provide a high-resolution, robust method to guide bacterial nomenclature that is aligned with classically obtained results. With the increasing availability of genome sequences, we sought to develop and apply a robust, portable, and high-resolution method for the assignment of genera and species designations that can recapitulate classically defined taxonomic designations. Using cutoffs derived from the lengths and sequence identities of core genome alignments along with phylogenetic analyses, we sought to evaluate or reevaluate genus- and species-level designations for diverse taxa, with an emphasis on the order , where species designations have been applied inconsistently. Our results indicate that the genus has an overabundance of species designations, that the current and genus designations are both too broad and need to be divided, and that there are clear demarcations of species that do not align precisely with the existing supergroup designations.
随着拥有基因组序列数据的细菌分类单元数量呈指数增长,需要一种新的标准化方法来指定物种名称,该方法应与传统获得的分类分析结果一致。对于不可培养的专性细胞内细菌来说,这一需求尤为迫切,因为无法使用传统定义的方法,如DNA-DNA杂交,来对它们进行分类,例如 中的那些细菌。在本研究中,我们为一系列具有传统定义物种的属以及 中的属生成了基于核苷酸的核心基因组比对。我们创建了一个工作流程,该流程利用从核心基因组比对中推断出的长度、序列同一性和系统发育关系来指定属和种的名称,使其与传统获得的结果相符。使用这种方法,大多数传统定义的细菌属都有一个核心基因组比对,其长度≥平均输入基因组长度的10%。 和 均未达到这一标准,这表明这些属的分类需要重新审视。一致的是,具有相同种加词的生物体的核心基因组比对具有≥96.8%的同一性。此外,这些核心基因组比对可用于生成系统发育基因组树,以识别定义物种的单系类群,并生成邻接网络树,以评估不同分类单元之间的重组情况。根据这些标准, 生物体被划分为与当前使用的超群指定不同的物种,而 生物体被划分为9个不同的物种,相比之下当前为27个物种。通过使用核心基因组比对来指定分类名称,我们旨在提供一种高分辨率、稳健的方法来指导细菌命名,使其与传统获得的结果一致。随着基因组序列的可用性不断提高,我们试图开发并应用一种稳健、便携且高分辨率的方法来指定属和种的名称,该方法能够重现传统定义的分类名称。利用从核心基因组比对的长度和序列同一性得出的阈值以及系统发育分析,我们试图评估或重新评估不同分类单元的属和种水平的名称,重点关注 目,在该目中物种名称的应用并不一致。我们的结果表明, 属的物种名称过多,当前的 和 属的指定都过于宽泛,需要进行划分,并且 物种存在明显的划分,与现有的超群指定并不完全一致。