APC Microbiome Ireland, University College Cork, Cork, Ireland.
School of Microbiology, University College Cork, Cork, Ireland.
BMC Genomics. 2018 Dec 14;19(1):931. doi: 10.1186/s12864-018-5313-6.
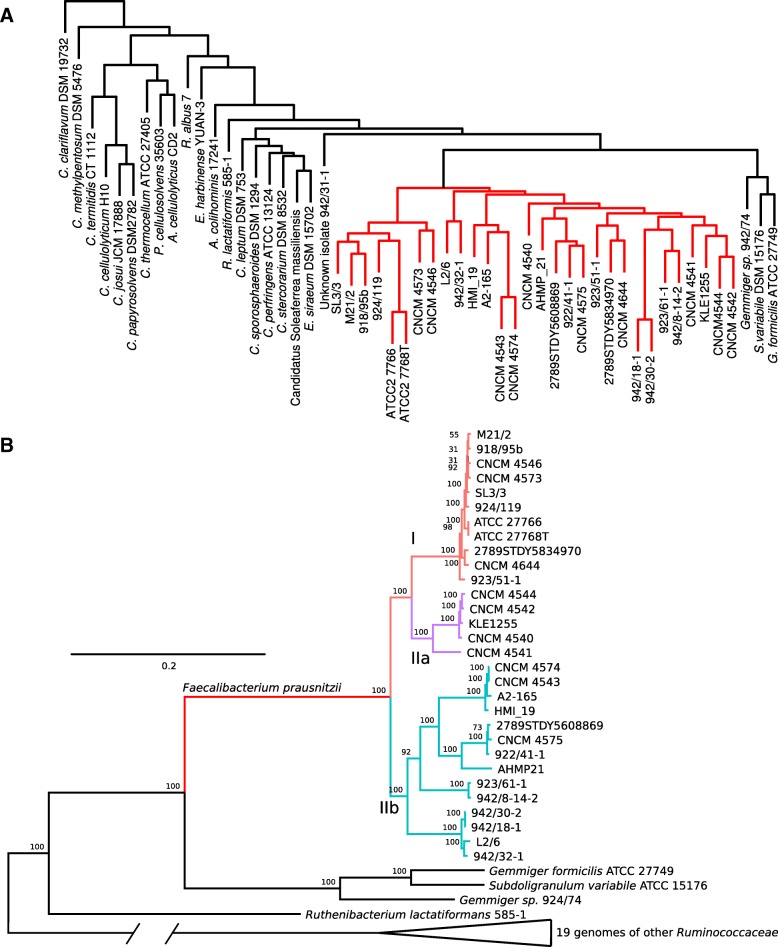
Faecalibacterium prausnitzii is a ubiquitous member of the human gut microbiome, constituting up to 15% of the total bacteria in the human gut. Substantial evidence connects decreased levels of F. prausnitzii with the onset and progression of certain forms of inflammatory bowel disease, which has been attributed to its anti-inflammatory potential. Two phylogroups of F. prausnitzii have been identified, with a decrease in phylogroup I being a more sensitive marker of intestinal inflammation. Much of the genomic and physiological data available to date was collected using phylogroup II strains. Little analysis of F. prausnitzii genomes has been performed so far and genetic differences between phylogroups I and II are poorly understood.
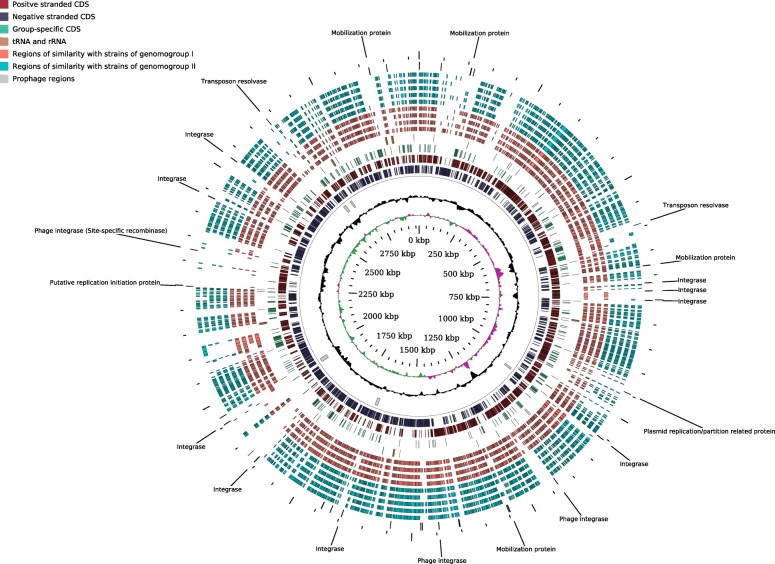
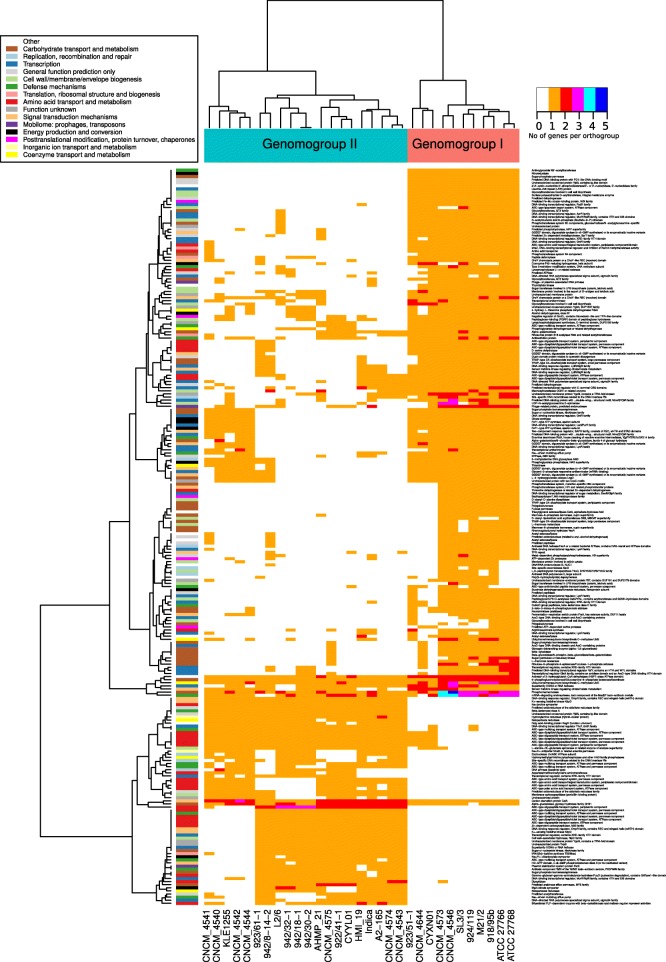
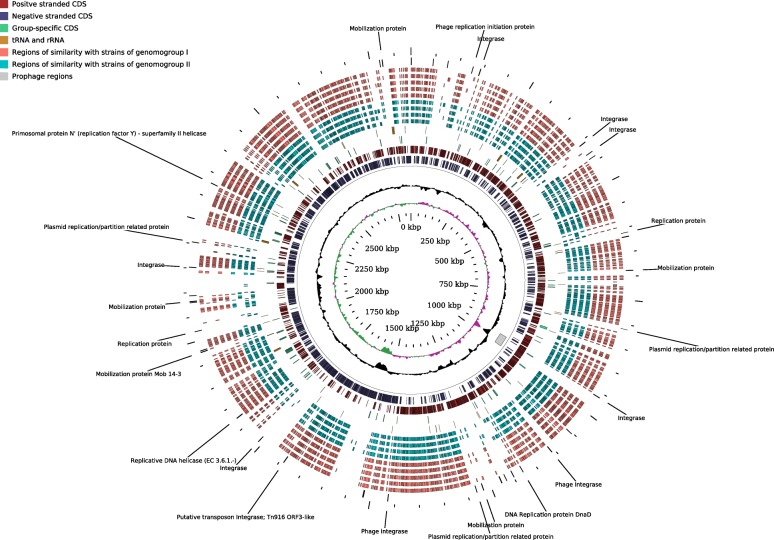
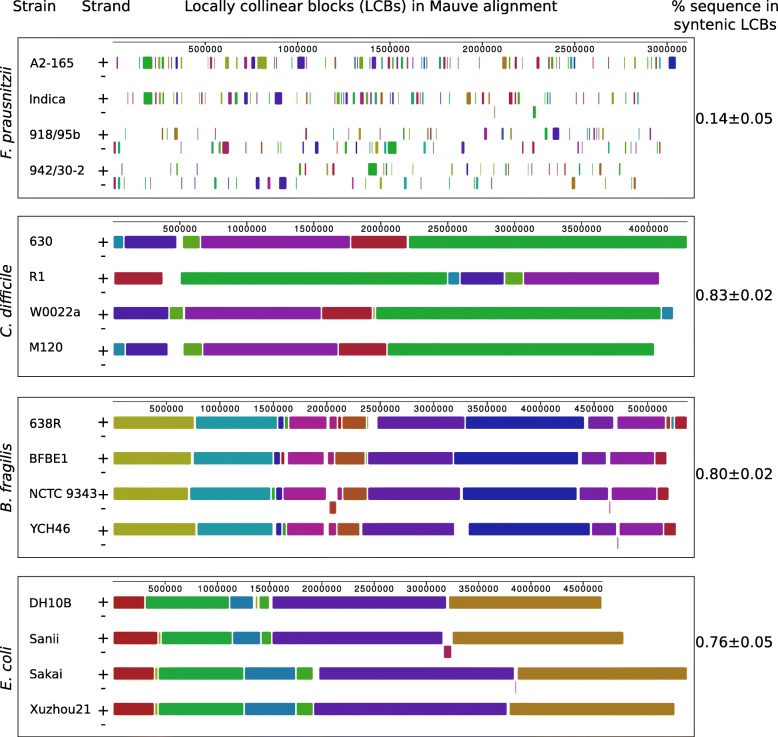
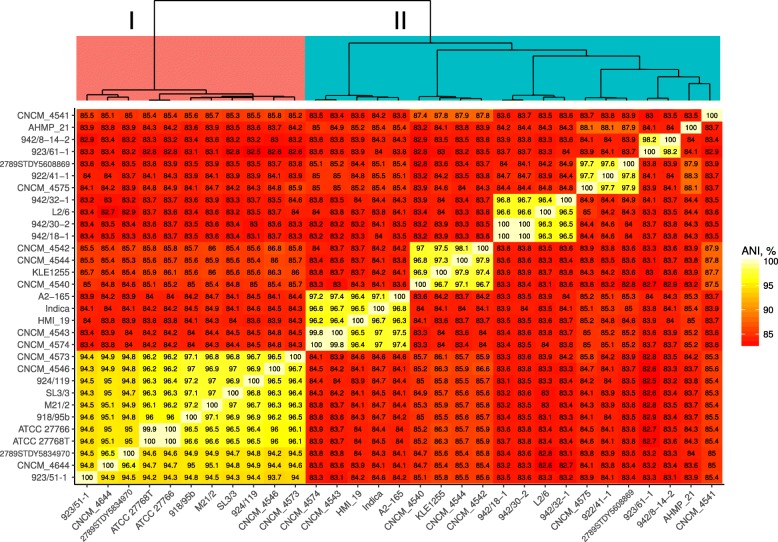
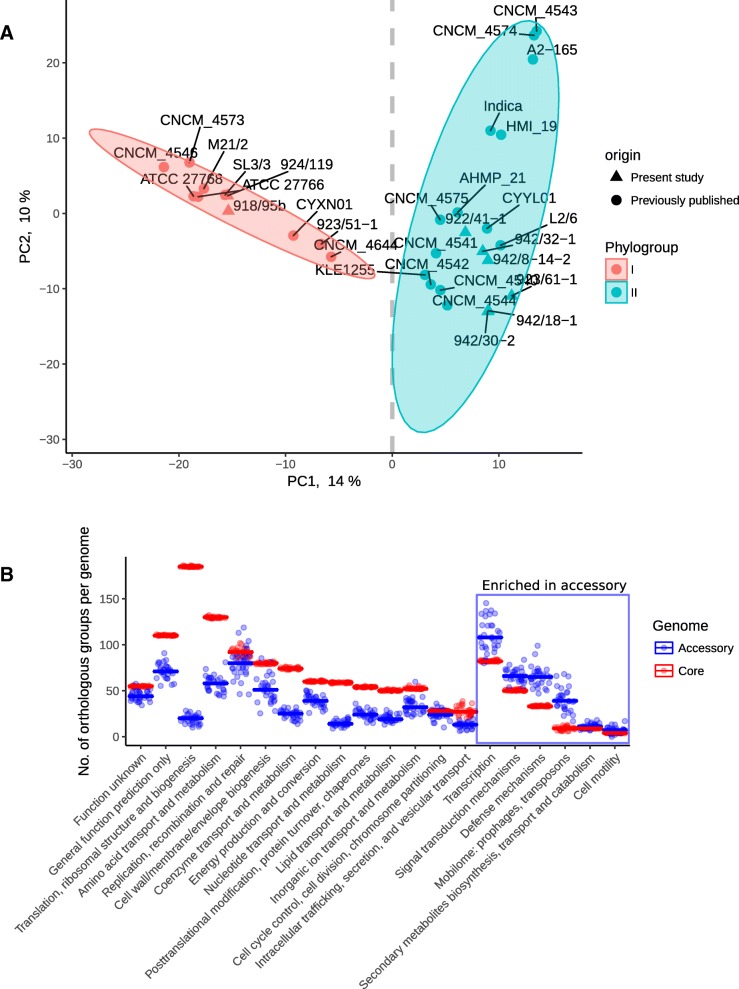
In this study we sequenced 11 additional F. prausnitzii genomes and performed comparative genomics to investigate intraspecies diversity, functional gene complement and the mobilome of 31 high-quality draft and complete genomes. We reveal a very low level of average nucleotide identity among F. prausnitzii genomes and a high level of genome plasticity. Two genomogroups can be separated based on differences in functional gene complement, albeit that this division does not fully agree with separation based on conserved gene phylogeny, highlighting the importance of horizontal gene transfer in shaping F. prausnitzii genomes. The difference between the two genomogroups is mainly in the complement of genes associated with catabolism of carbohydrates (such as a predicted sialidase gene in genomogroup I) and amino acids, as well as defense mechanisms.
Based on the combination of ANI of genomic sequences, phylogenetic analysis of core proteomes and functional differences we propose to separate the species F. prausnitzii into two new species level taxa: F. prausnitzii sensu stricto (neotype strain A2-165 = DSM 17677 = JCM 31915) and F. moorei sp. nov. (type strain ATCC 27768 = NCIMB 13872).
普拉梭菌是人类肠道微生物群中无处不在的成员,构成了人类肠道中总细菌的 15%。大量证据表明,普拉梭菌水平的降低与某些形式的炎症性肠病的发生和进展有关,这归因于其抗炎潜力。已鉴定出普拉梭菌的两个系统发育群,I 群的减少是肠道炎症更敏感的标志物。迄今为止,可用的大部分基因组和生理数据都是使用 II 群菌株收集的。到目前为止,对普拉梭菌基因组的分析很少,I 群和 II 群之间的遗传差异也知之甚少。
在这项研究中,我们对 11 株额外的普拉梭菌基因组进行了测序,并进行了比较基因组学分析,以研究 31 株高质量草图和完整基因组的种内多样性、功能基因组成和可移动基因组。我们揭示了普拉梭菌基因组之间平均核苷酸同一性非常低,基因组可塑性很高。可以根据功能基因组成的差异将两个基因组群分开,尽管这种划分不完全与基于保守基因系统发育的分离一致,这突出了水平基因转移在塑造普拉梭菌基因组方面的重要性。两个基因组群之间的差异主要在于与碳水化合物(如 I 群中预测的唾液酸酶基因)和氨基酸以及防御机制的代谢相关的基因组成。
基于基因组序列的 ANI 组合、核心蛋白质组的系统发育分析和功能差异,我们建议将物种普拉梭菌分为两个新的种水平分类群:普拉梭菌(模式菌株 A2-165=DSM 17677=JCM 31915)和摩氏普拉梭菌。(模式菌株 ATCC 27768=NCIMB 13872)。