Yang Mary Qu, Weissman Sherman M, Yang William, Zhang Jialing, Canaann Allon, Guan Renchu
Joint Bioinformatics Program, University of Arkansas Little Rock George Washington Donaghey College of Engineering & IT and University of Arkansas for Medical Sciences, Little Rock, AR, 72204, USA.
Department of Genetics, Yale University, New Haven, CT, 06512, USA.
BMC Syst Biol. 2018 Dec 14;12(Suppl 7):114. doi: 10.1186/s12918-018-0638-y.
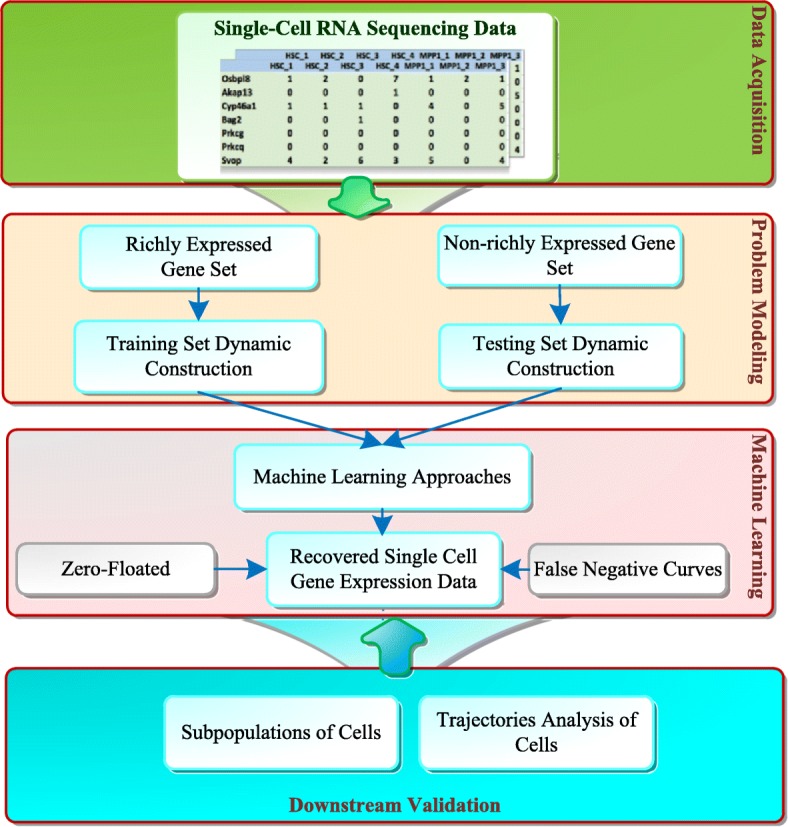
Single-cell RNA sequencing (scRNA-seq) technology provides an effective way to study cell heterogeneity. However, due to the low capture efficiency and stochastic gene expression, scRNA-seq data often contains a high percentage of missing values. It has been showed that the missing rate can reach approximately 30% even after noise reduction. To accurately recover missing values in scRNA-seq data, we need to know where the missing data is; how much data is missing; and what are the values of these data.
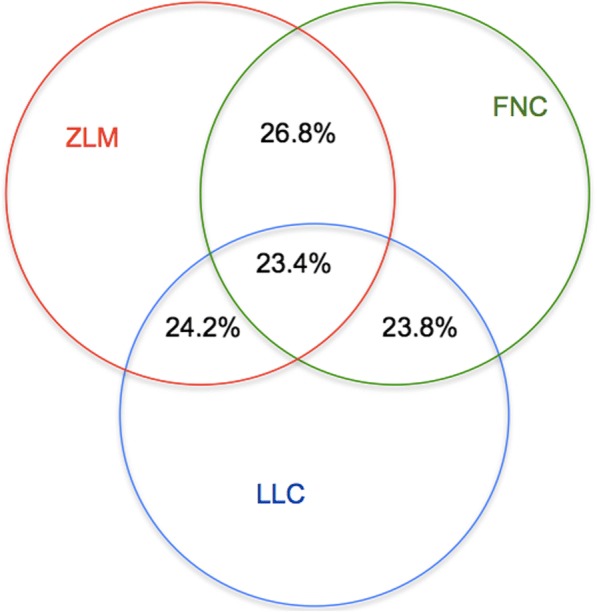
To solve these three problems, we propose a novel model with a hybrid machine learning method, namely, missing imputation for single-cell RNA-seq (MISC). To solve the first problem, we transformed it to a binary classification problem on the RNA-seq expression matrix. Then, for the second problem, we searched for the intersection of the classification results, zero-inflated model and false negative model results. Finally, we used the regression model to recover the data in the missing elements.
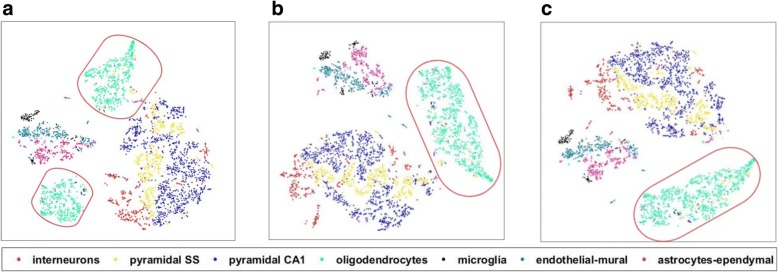
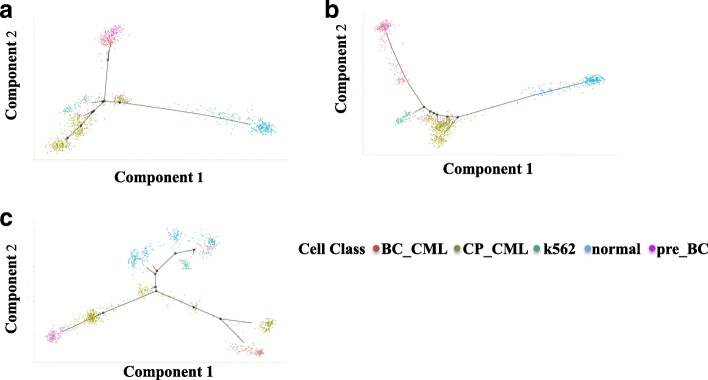
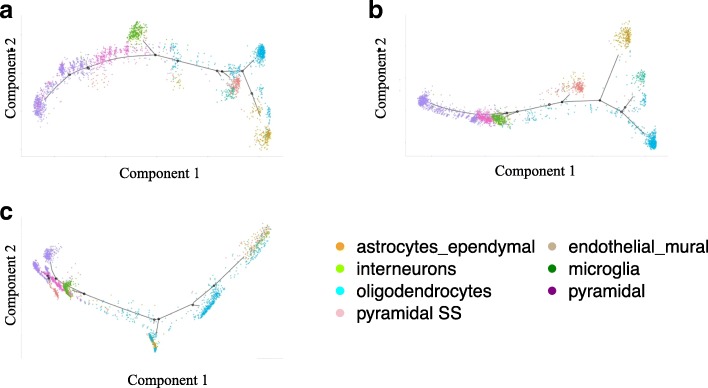
We compared the raw data without imputation, the mean-smooth neighbor cell trajectory, MISC on chronic myeloid leukemia data (CML), the primary somatosensory cortex and the hippocampal CA1 region of mouse brain cells. On the CML data, MISC discovered a trajectory branch from the CP-CML to the BC-CML, which provides direct evidence of evolution from CP to BC stem cells. On the mouse brain data, MISC clearly divides the pyramidal CA1 into different branches, and it is direct evidence of pyramidal CA1 in the subpopulations. In the meantime, with MISC, the oligodendrocyte cells became an independent group with an apparent boundary.
Our results showed that the MISC model improved the cell type classification and could be instrumental to study cellular heterogeneity. Overall, MISC is a robust missing data imputation model for single-cell RNA-seq data.
单细胞RNA测序(scRNA-seq)技术为研究细胞异质性提供了一种有效方法。然而,由于捕获效率低和基因表达的随机性,scRNA-seq数据通常包含高比例的缺失值。研究表明,即使经过降噪处理,缺失率仍可达到约30%。为了准确恢复scRNA-seq数据中的缺失值,我们需要知道缺失数据的位置;缺失了多少数据;以及这些数据的值是什么。
为了解决这三个问题,我们提出了一种采用混合机器学习方法的新型模型,即单细胞RNA-seq缺失值插补(MISC)。为了解决第一个问题,我们将其转化为RNA-seq表达矩阵上的二元分类问题。然后,对于第二个问题,我们寻找分类结果、零膨胀模型和假阴性模型结果的交集。最后利用回归模型恢复缺失元素中的数据。
我们比较了未插补的原始数据、平均平滑的相邻细胞轨迹、慢性髓系白血病数据(CML)、小鼠脑初级体感皮层和海马CA1区细胞上的MISC。在CML数据上,MISC发现了一条从慢性期慢性髓系白血病(CP-CML)到急变期慢性髓系白血病(BC-CML)的轨迹分支,这为从CP干细胞向BC干细胞的进化提供了直接证据。在小鼠脑数据上,MISC清楚地将锥体CA1细胞分为不同分支,这是锥体CA1细胞亚群的直接证据。同时,使用MISC后,少突胶质细胞成为一个具有明显边界的独立组。
我们的结果表明,MISC模型改善了细胞类型分类,有助于研究细胞异质性。总体而言,MISC是一种用于单细胞RNA-seq数据的强大缺失数据插补模型。