Systems Biology Initiative, School of Biotechnology and Biomolecular Sciences, UNSW Sydney, Sydney, NSW, 2052, Australia.
Gastrointestinal and Liver Unit, The Prince of Wales Hospital, Randwick, NSW, 2031, Australia.
Microbiome. 2018 Dec 17;6(1):227. doi: 10.1186/s40168-018-0611-4.
The esophageal microbiome has been proposed to be involved in a range of diseases including the esophageal adenocarcinoma cascade; however, little is currently known about its function and relationship to the host. Here, the esophageal microbiomes of 106 prospectively recruited patients were assessed using 16S rRNA and 18S rRNA amplicon sequencing as well as shotgun sequencing, and associations with age, gender, proton pump inhibitor use, host genetics, and disease were tested.
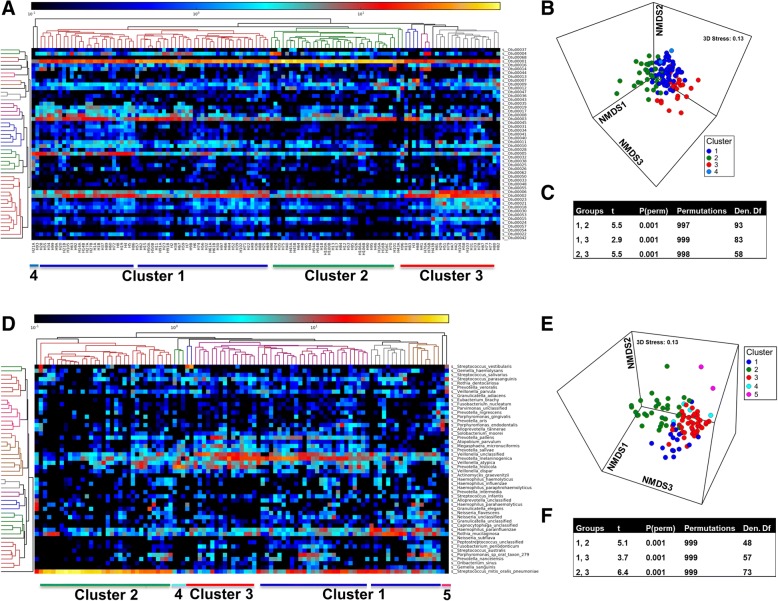
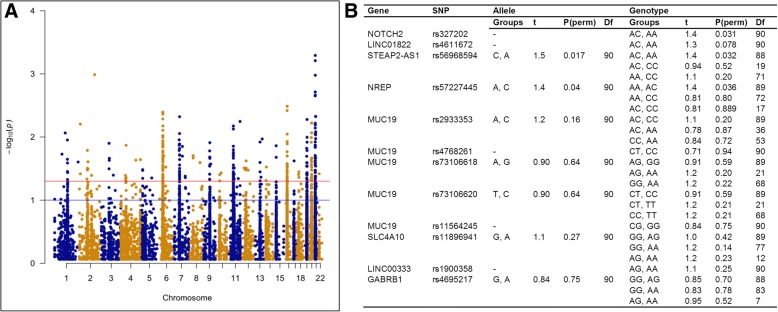
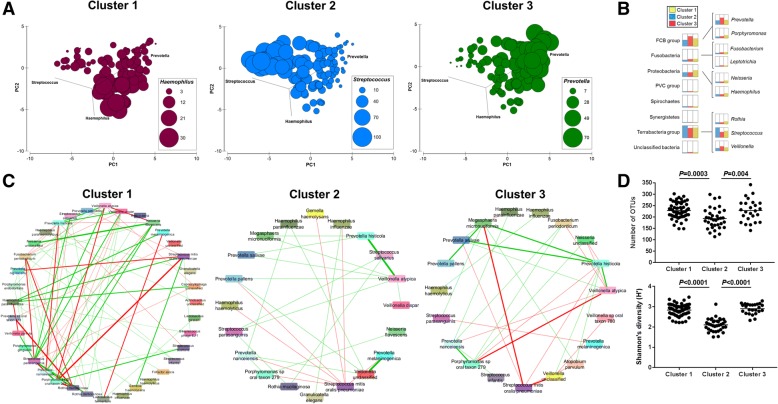
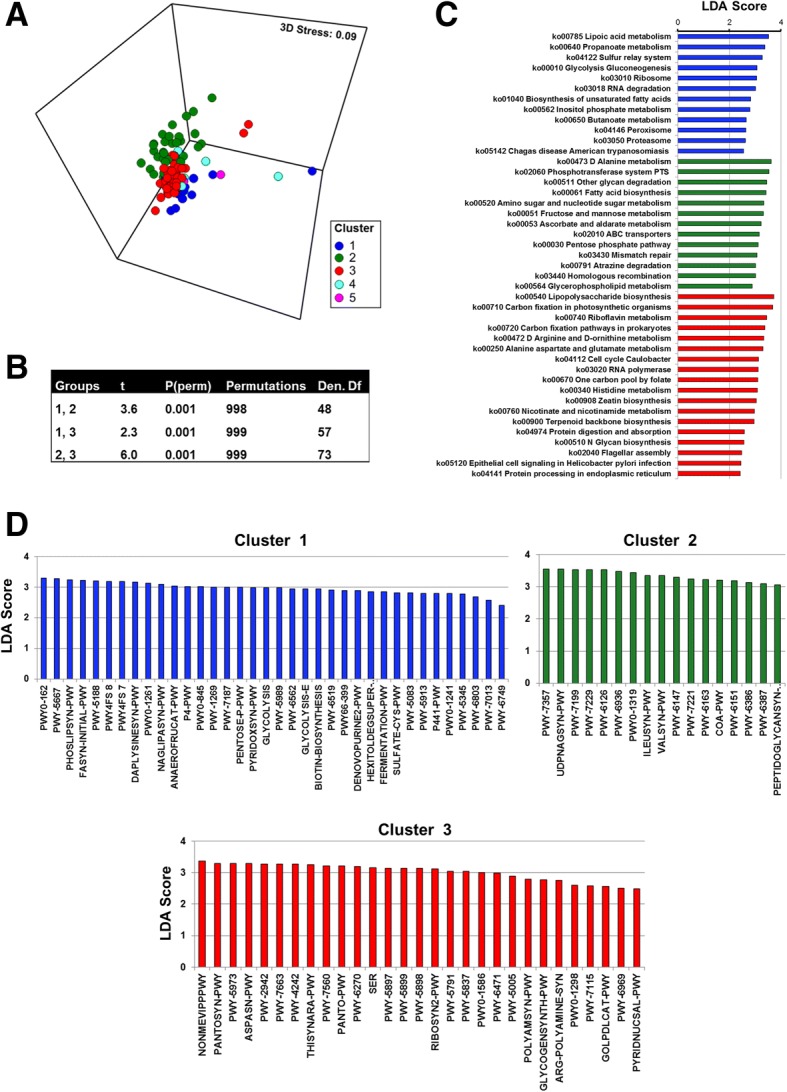
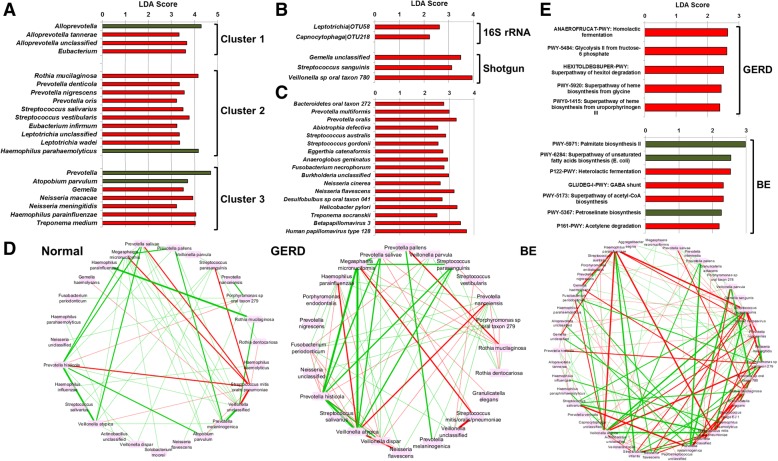
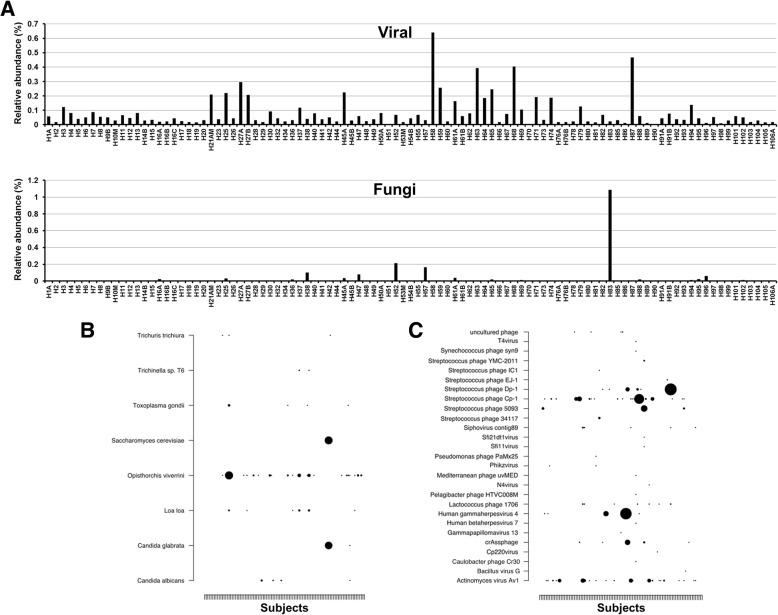
The esophageal microbiome was found to cluster into functionally distinct community types (esotypes) defined by the relative abundances of Streptococcus and Prevotella. While age was found to be a significant factor driving microbiome composition, bacterial signatures and functions such as enrichment with Gram-negative oral-associated bacteria and microbial lactic acid production were associated with the early stages of the esophageal adenocarcinoma cascade. Non-bacterial microbes such as archaea, Candida spp., and bacteriophages were also identified in low abundance in the esophageal microbiome. Specific host SNPs in NOTCH2, STEAP2-AS1, and NREP were associated with the composition of the esophageal microbiome in our cohort.
This study provides the most comprehensive assessment of the esophageal microbiome to date and identifies novel signatures and host markers that can be investigated further in the context of esophageal adenocarcinoma development.
食管微生物组被认为与多种疾病有关,包括食管腺癌级联反应;然而,目前对于其功能及其与宿主的关系知之甚少。在这里,通过 16S rRNA 和 18S rRNA 扩增子测序以及鸟枪法测序,对 106 名前瞻性招募的患者的食管微生物组进行了评估,并测试了与年龄、性别、质子泵抑制剂使用、宿主遗传学和疾病的关联。
发现食管微生物组通过链球菌和普雷沃氏菌相对丰度聚类为具有不同功能的群落类型(esotypes)。尽管年龄被发现是驱动微生物组组成的重要因素,但细菌特征和功能,如革兰氏阴性口腔相关细菌的富集和微生物乳酸生产,与食管腺癌级联反应的早期阶段有关。在食管微生物组中也以低丰度鉴定出非细菌微生物,如古菌、念珠菌属和噬菌体。NOTCH2、STEAP2-AS1 和 NREP 中的特定宿主 SNP 与我们队列中食管微生物组的组成有关。
本研究提供了迄今为止对食管微生物组最全面的评估,并确定了可在食管腺癌发展背景下进一步研究的新特征和宿主标志物。