Research Unit in Bioinformatics (RUBi), Department of Biochemistry and Microbiology, Rhodes University, Grahamstown, 6140, South Africa.
Department of Mathematics (Pure & Applied), Rhodes University, Grahamstown, 6140, South Africa.
Sci Rep. 2018 Dec 18;8(1):17938. doi: 10.1038/s41598-018-36041-8.
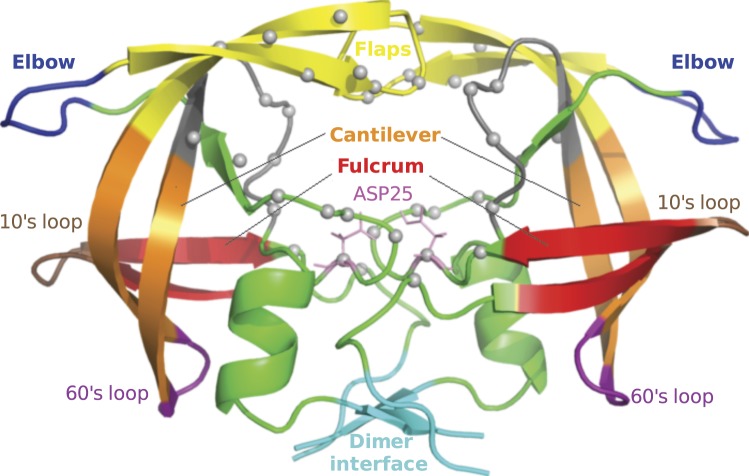
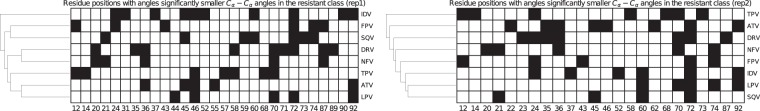
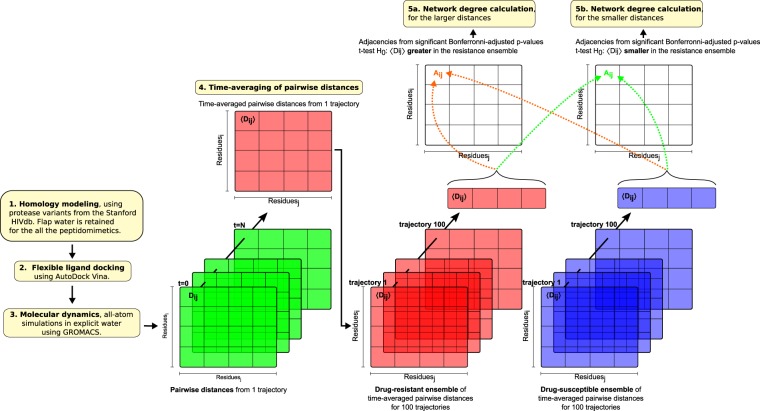
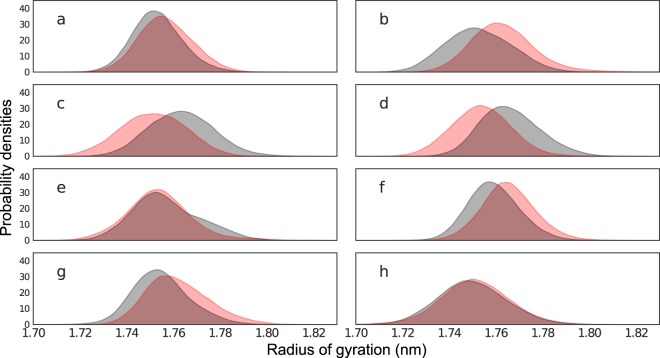
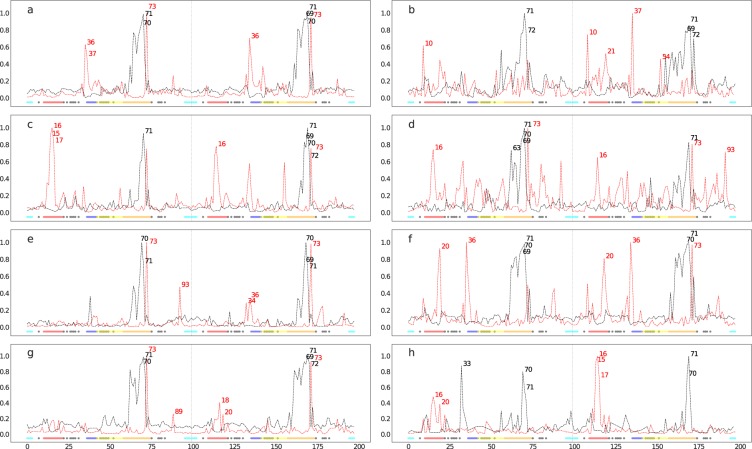
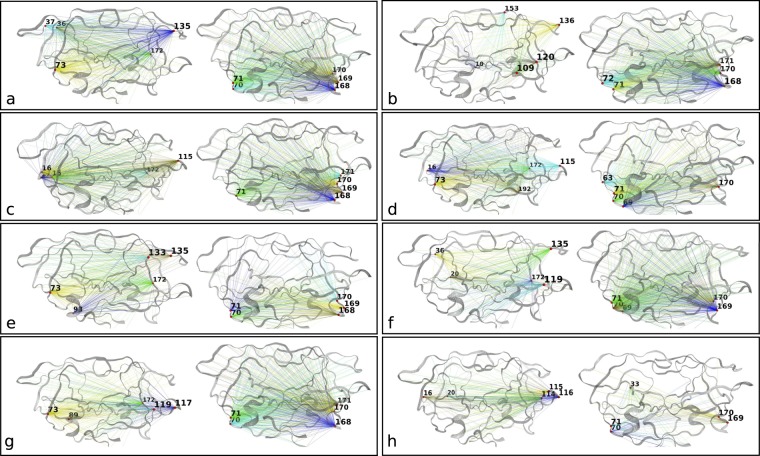
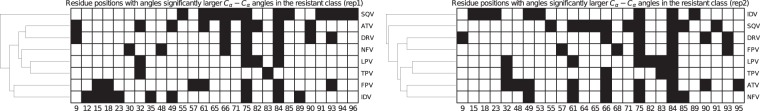
The use of antiretrovirals (ARVs) has drastically improved the life quality and expectancy of HIV patients since their introduction in health care. Several millions are still afflicted worldwide by HIV and ARV resistance is a constant concern for both healthcare practitioners and patients, as while treatment options are finite, the virus constantly adapts via complex mutation patterns to select for resistant strains under the pressure of drug treatment. The HIV protease is a crucial enzyme for viral maturation and has been a game changing drug target since the first application. Due to similarities in protease inhibitor designs, drug cross-resistance is not uncommon across ARVs of the same class. It is known that resistance against protease inhibitors is associated with a wider active site, but results from our large scale molecular dynamics simulations combined with statistical tests and network analysis further show, for the first time, that there are regions of local expansions and compactions associated with high levels of resistance conserved across eight different protease inhibitors visible in their complexed form within closed receptor conformations. The observed conserved expansion sites may provide an alternative drug-targeting site. Further, the method developed here is novel, supplementary to methods of variation analysis at sequence level, and should be applicable in analysing the structural consequences of mutations in other contexts using molecular ensembles.
抗逆转录病毒药物(ARVs)的使用自引入医疗保健领域以来,极大地提高了 HIV 患者的生活质量和预期寿命。全世界仍有数百万人受到 HIV 的影响,而 ARV 耐药性是医疗保健从业者和患者一直关注的问题,因为虽然治疗选择有限,但病毒通过复杂的突变模式不断适应,在药物治疗的压力下选择耐药株。HIV 蛋白酶是病毒成熟的关键酶,自首次应用以来,它一直是改变游戏规则的药物靶点。由于蛋白酶抑制剂设计的相似性,同一类 ARV 之间药物交叉耐药性并不罕见。已知蛋白酶抑制剂的耐药性与更宽的活性位点有关,但我们的大规模分子动力学模拟结合统计测试和网络分析的结果首次表明,在封闭受体构象中,其复合物形式下可见的八种不同蛋白酶抑制剂中存在与高水平耐药性相关的局部扩展和压缩区域。观察到的保守扩展位点可能提供了一个替代的药物靶点。此外,这里开发的方法是新颖的,补充了序列水平上的变异分析方法,并且应该适用于使用分子集合分析其他情况下突变的结构后果。