The Institute of Cancer Research , Chester Beatty Laboratories, London SW3 6JB , U.K.
Wellcome Sanger Institute , Wellcome Genome Campus , Cambridge CB10 1SA , U.K.
J Proteome Res. 2019 Mar 1;18(3):1433-1440. doi: 10.1021/acs.jproteome.8b00870. Epub 2019 Jan 3.
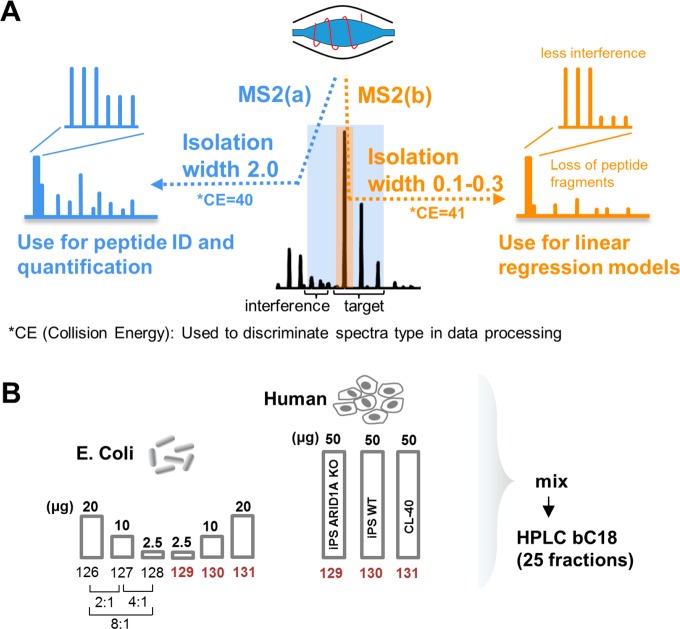
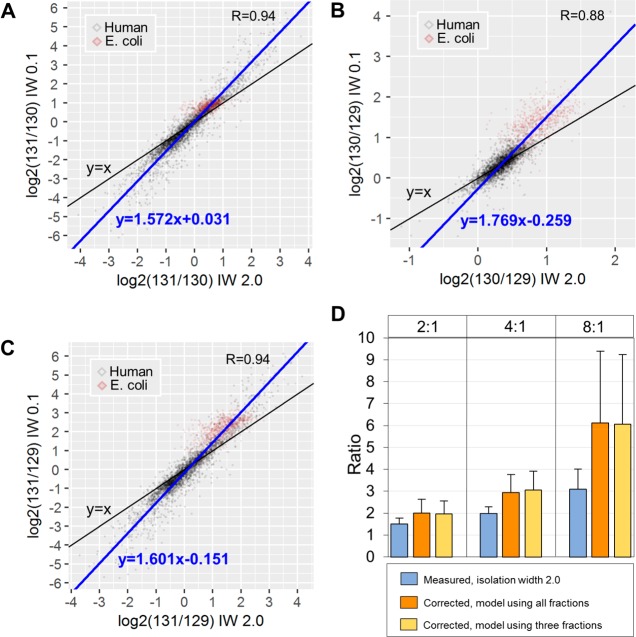
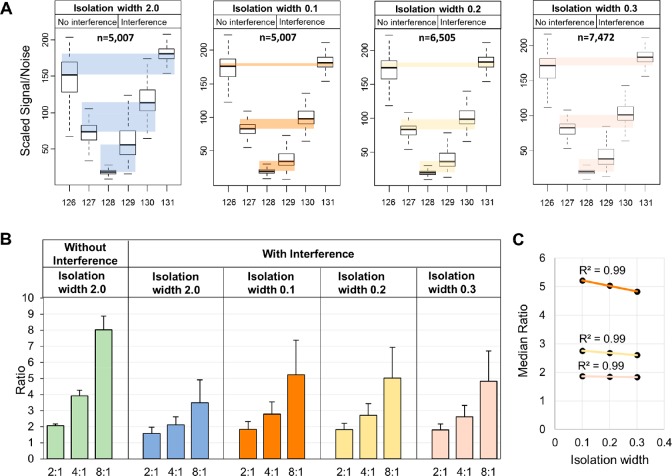
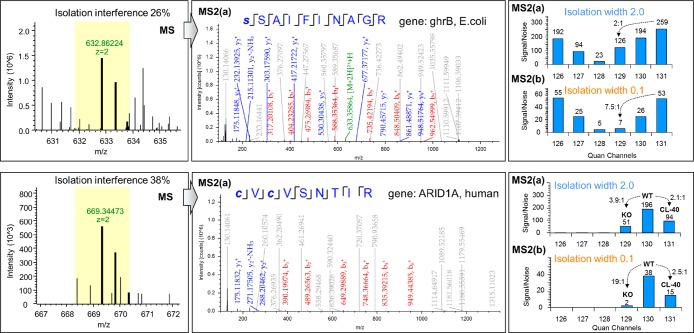
Isobaric labeling is a highly precise approach for protein quantification. However, due to the isolation interference problem, isobaric tagging suffers from ratio underestimation at the MS2 level. The use of narrow isolation widths is a rational approach to alleviate the interference problem; however, this approach compromises proteome coverage. We reasoned that although a very narrow isolation window will result in loss of peptide fragment ions, the reporter ion signals will be retained for a significant portion of the spectra. On the basis of this assumption, we have designed a dual isolation width acquisition (DIWA) method, in which each precursor is first fragmented with HCD using a standard isolation width for peptide identification and preliminary quantification, followed by a second MS2 HCD scan using a much narrower isolation width for the acquisition of quantitative spectra with reduced interference. We leverage the quantification obtained by the "narrow" scans to build linear regression models and apply these to decompress the fold-changes measured at the "standard" scans. We evaluate the DIWA approach using a nested two species/gene knockout TMT-6plex experimental design and discuss the perspectives of this approach.
等压标记是一种高度精确的蛋白质定量方法。然而,由于分离干扰问题,等压标记在 MS2 水平上存在比值低估的问题。使用较窄的隔离宽度是缓解干扰问题的合理方法;然而,这种方法会影响蛋白质组覆盖率。我们推断,尽管非常窄的隔离窗口会导致肽片段离子的丢失,但报告离子信号将在很大一部分光谱中保留。基于这一假设,我们设计了一种双隔离宽度采集(DIWA)方法,其中每个前体首先使用标准隔离宽度进行 HCD 碎裂,用于肽鉴定和初步定量,然后使用更窄的隔离宽度进行第二次 MS2 HCD 扫描,以获取干扰较小的定量谱。我们利用“窄”扫描获得的定量信息来构建线性回归模型,并将这些模型应用于解压缩“标准”扫描测量的折叠变化。我们使用嵌套的两种物种/基因敲除 TMT-6plex 实验设计来评估 DIWA 方法,并讨论该方法的前景。