School of Life Sciences, Tsinghua University, Beijing 100084, China; State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences(Beijing), Beijing Institute of Lifeomics, Beijing 102206, China.
Department of Biomedical Informatics, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China; Institute of Systems Biomedicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China.
EBioMedicine. 2019 Feb;40:305-317. doi: 10.1016/j.ebiom.2018.12.039. Epub 2018 Dec 26.
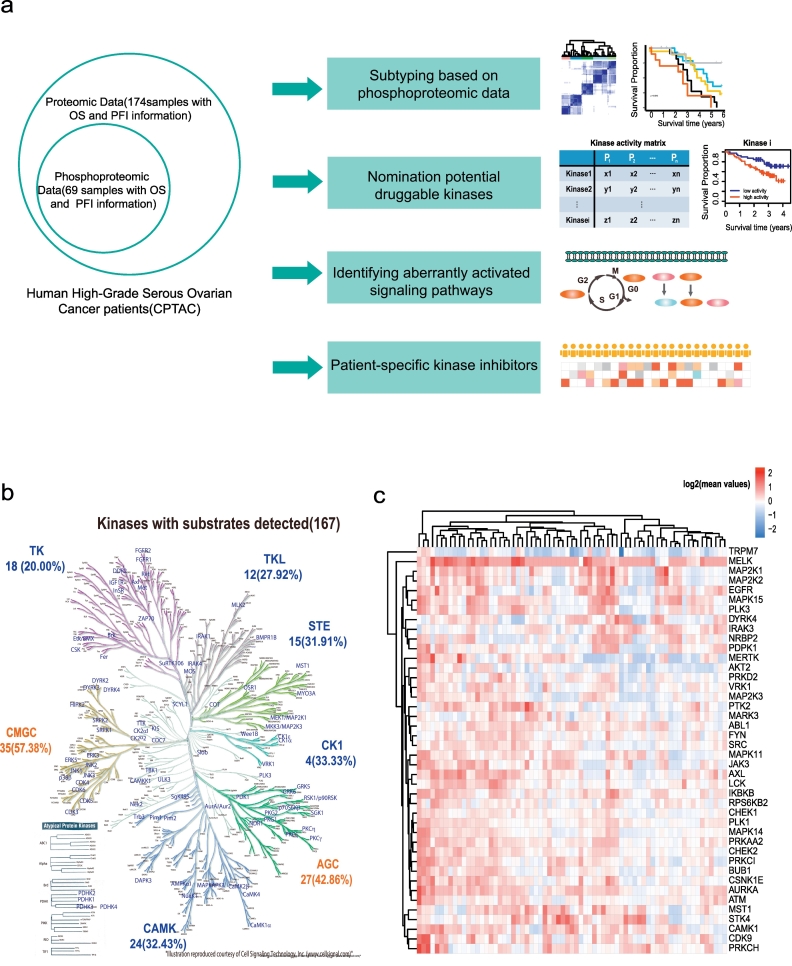
Molecular subtyping of cancer aimed to predict patient overall survival (OS) and nominate drug targets for patient treatments is central to precision oncology. Owing to the rapid development of phosphoproteomics, we can now measure thousands of phosphoproteins in human cancer tissues. However, limited studies report how to analyse the complex phosphoproteomic data for cancer subtyping and to nominate druggable kinase candidates.
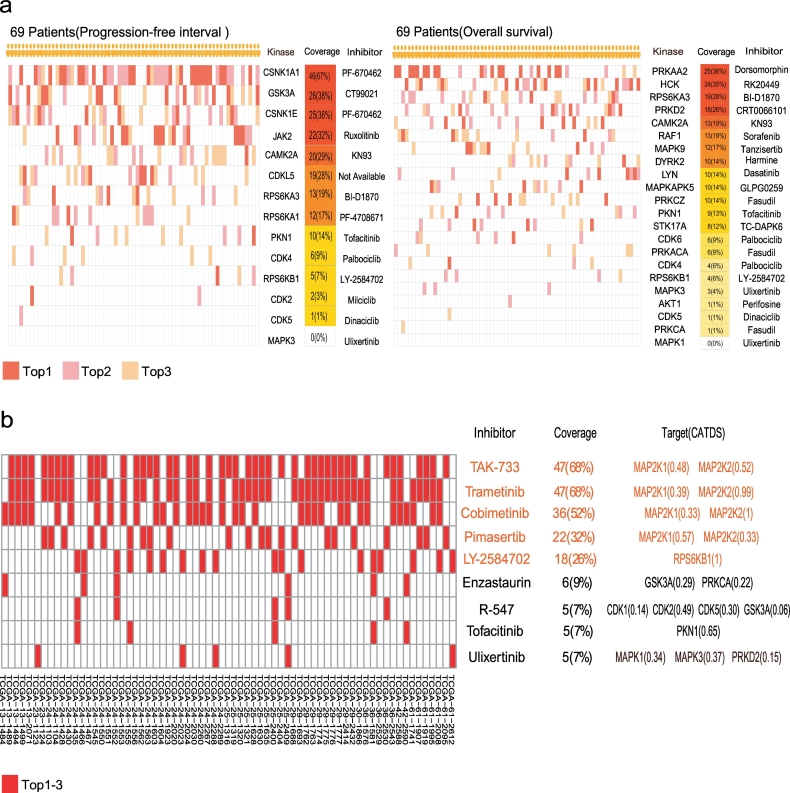
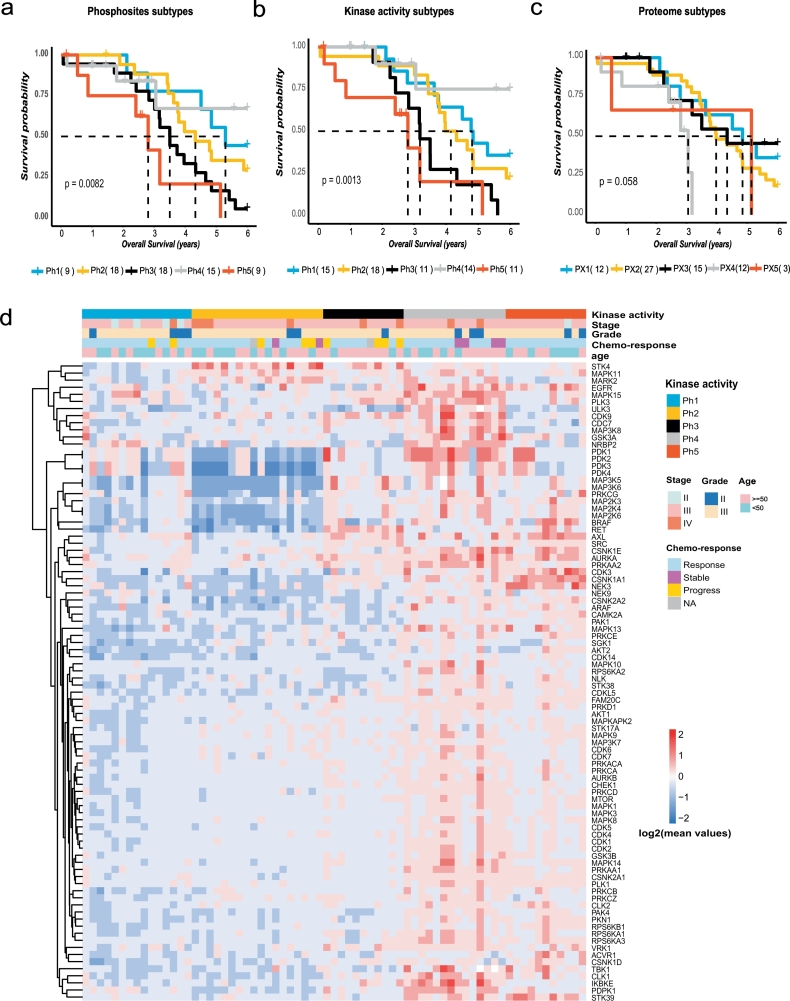
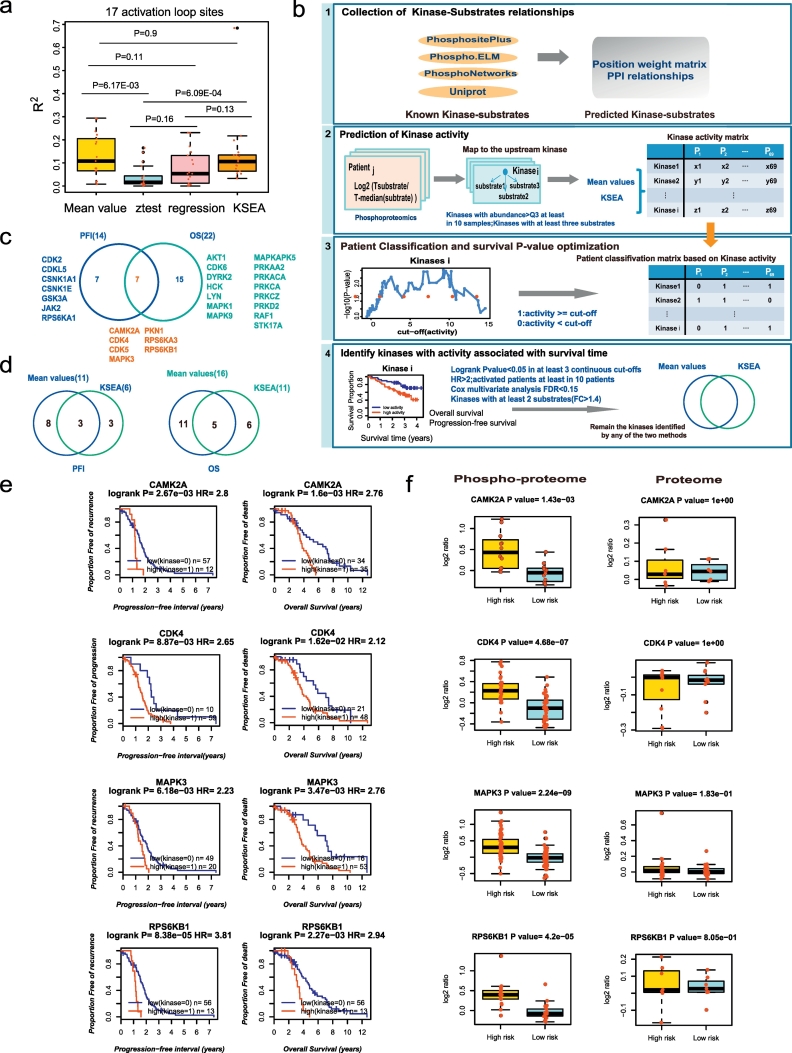
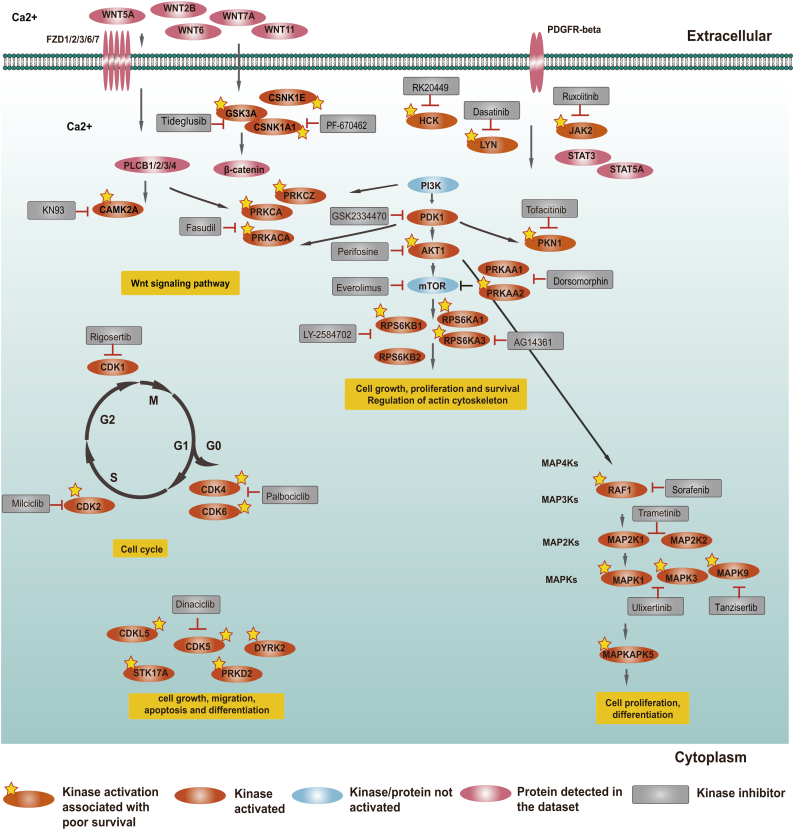
In this work, we reanalysed the phosphoproteomic data of high-grade serous ovarian cancer (HGSOC) from the Clinical Proteomic Tumour Analysis Consortium (CPTAC). Our analysis classified HGSOC into 5 major subtypes that were associated with different OS and appeared to be more accurate than that achieved with protein profiling. We provided a workflow to identify 29 kinases whose increased activities in tumours are associated with poor survival. The altered kinase signalling landscape of HGSOC included the PI3K/AKT/mTOR, cell cycle and MAP kinase signalling pathways. We also developed a "patient-specific" hierarchy of clinically actionable kinases and selected kinase inhibitors by considering kinase activation and kinase inhibitor selectivity.
Our study offered a global phosphoproteomics data analysis workflow to aid in cancer molecular subtyping, determining phosphorylation-based cancer hallmarks and facilitating nomination of kinase inhibition in cancer.
癌症的分子亚型分析旨在预测患者的总生存期(OS),并为患者治疗指定药物靶点,这是精准肿瘤学的核心。由于磷酸化蛋白质组学的快速发展,我们现在可以在人类癌症组织中测量数千种磷酸化蛋白。然而,有限的研究报告了如何分析癌症亚型分析和可用药激酶候选物的复杂磷酸化蛋白质组数据。
在这项工作中,我们重新分析了临床蛋白质组肿瘤分析联盟(CPTAC)的高级别浆液性卵巢癌(HGSOC)的磷酸蛋白质组数据。我们的分析将 HGSOC 分为 5 个主要亚型,这些亚型与不同的 OS 相关,并且似乎比蛋白质谱分析更准确。我们提供了一种工作流程来识别 29 种激酶,其在肿瘤中的活性增加与生存率降低相关。HGSOC 的改变的激酶信号转导景观包括 PI3K/AKT/mTOR、细胞周期和 MAP 激酶信号通路。我们还通过考虑激酶激活和激酶抑制剂选择性,开发了一种“患者特异性”的临床可操作激酶层次结构,并选择了激酶抑制剂。
我们的研究提供了一种全局磷酸蛋白质组学数据分析工作流程,以帮助癌症分子亚型分析、确定基于磷酸化的癌症特征,并促进癌症中激酶抑制的提名。