Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA.
BMC Genomics. 2019 Jan 9;20(1):23. doi: 10.1186/s12864-018-5381-7.
Short-read sequencing technologies have made microbial genome sequencing cheap and accessible. However, closing genomes is often costly and assembling short reads from genomes that are repetitive and/or have extreme %GC content remains challenging. Long-read, single-molecule sequencing technologies such as the Oxford Nanopore MinION have the potential to overcome these difficulties, although the best approach for harnessing their potential remains poorly evaluated.
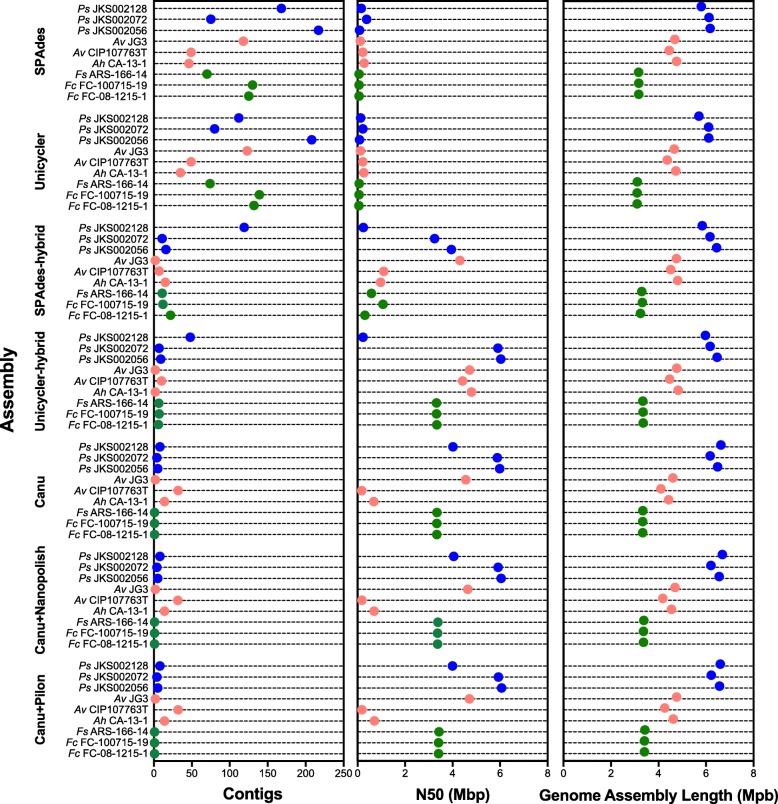
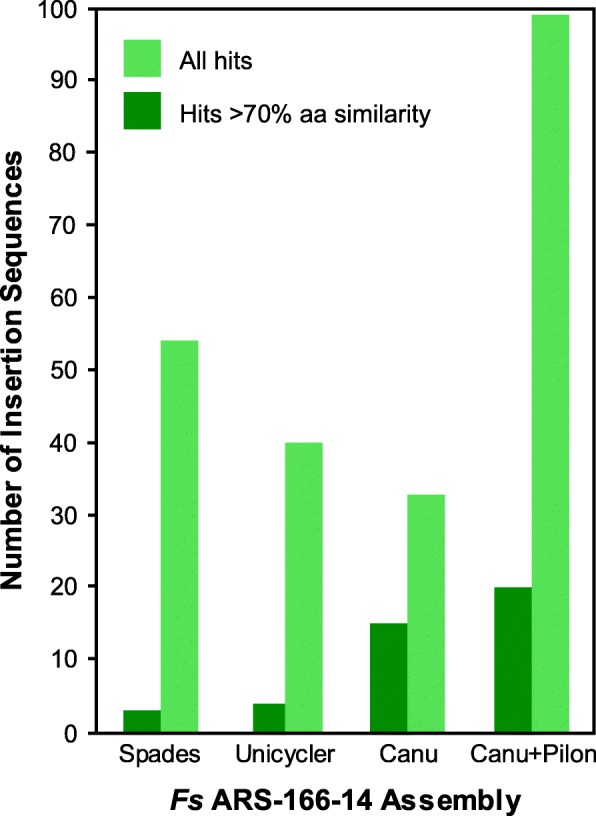
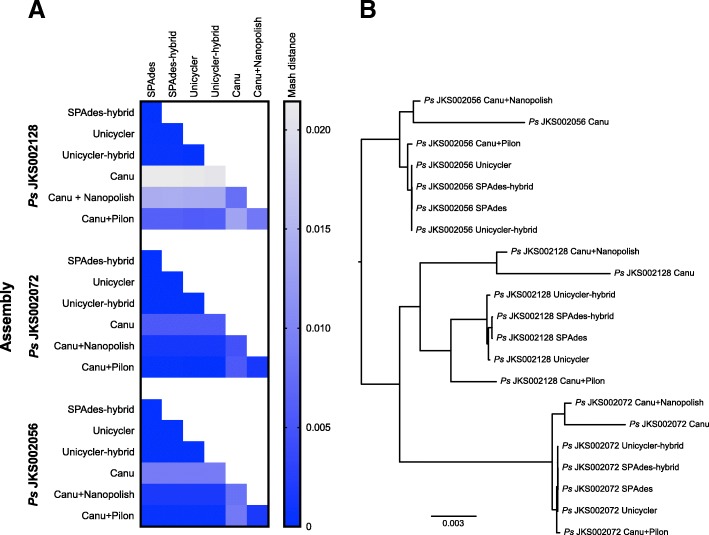
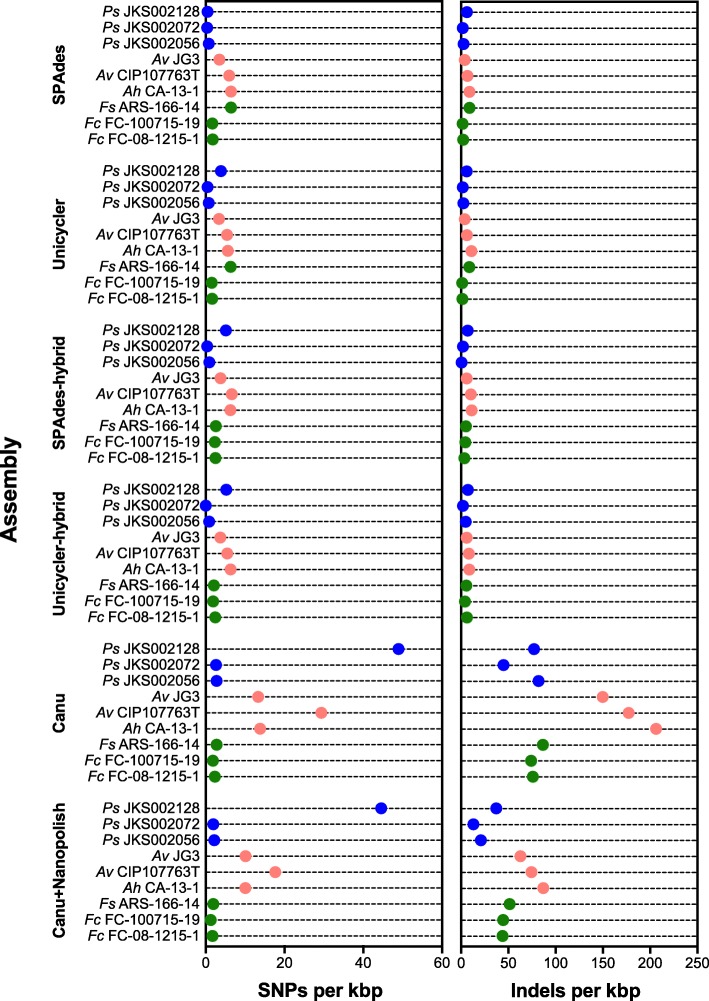
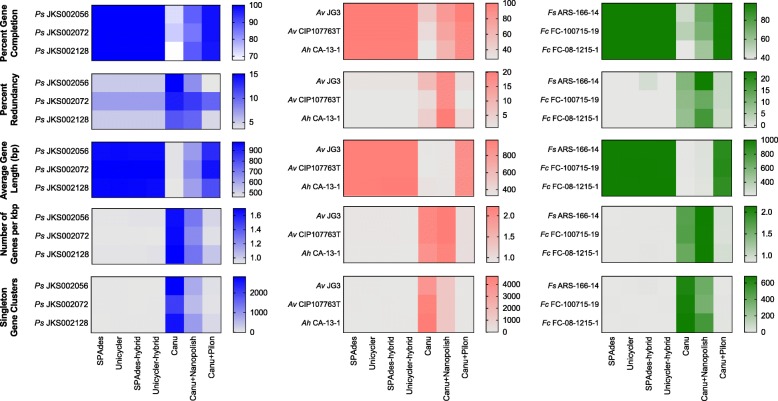
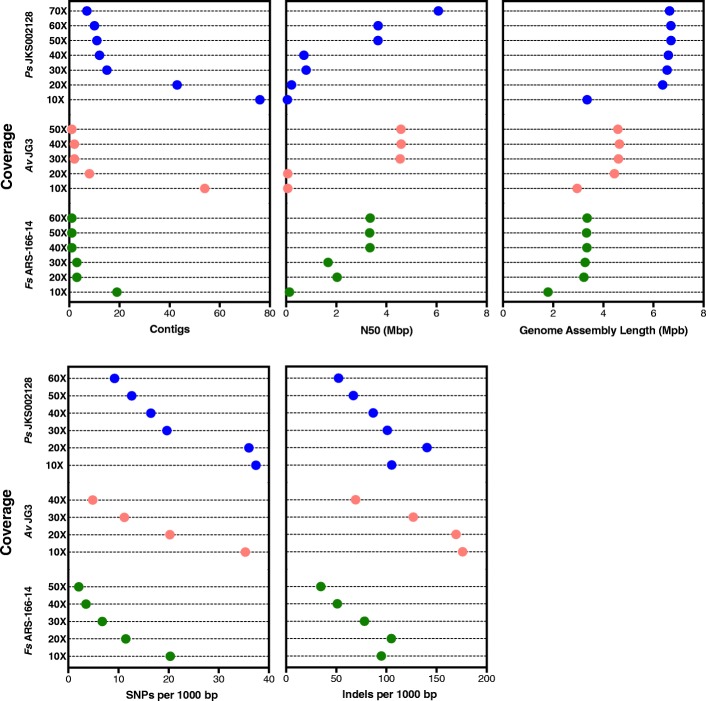
We sequenced nine bacterial genomes spanning a wide range of GC contents using Illumina MiSeq and Oxford Nanopore MinION sequencing technologies to determine the advantages of each approach, both individually and combined. Assemblies using only MiSeq reads were highly accurate but lacked contiguity, a deficiency that was partially overcome by adding MinION reads to these assemblies. Even more contiguous genome assemblies were generated by using MinION reads for initial assembly, but these assemblies were more error-prone and required further polishing. This was especially pronounced when Illumina libraries were biased, as was the case for our strains with both high and low GC content. Increased genome contiguity dramatically improved the annotation of insertion sequences and secondary metabolite biosynthetic gene clusters, likely because long-reads can disambiguate these highly repetitive but biologically important genomic regions.
Genome assembly using short-reads is challenged by repetitive sequences and extreme GC contents. Our results indicate that these difficulties can be largely overcome by using single-molecule, long-read sequencing technologies such as the Oxford Nanopore MinION. Using MinION reads for assembly followed by polishing with Illumina reads generated the most contiguous genomes with sufficient accuracy to enable the accurate annotation of important but difficult to sequence genomic features such as insertion sequences and secondary metabolite biosynthetic gene clusters. The combination of Oxford Nanopore and Illumina sequencing can therefore cost-effectively advance studies of microbial evolution and genome-driven drug discovery.
短读测序技术使微生物基因组测序变得廉价且易于实现。然而,基因组的闭合通常成本高昂,并且组装具有重复序列和/或极端 GC 含量的基因组的短读序列仍然具有挑战性。长读长、单分子测序技术,如牛津纳米孔 MinION,具有克服这些困难的潜力,尽管利用其潜力的最佳方法仍未得到很好的评估。
我们使用 Illumina MiSeq 和牛津纳米孔 MinION 测序技术对跨越广泛 GC 含量范围的九个细菌基因组进行测序,以确定每种方法(单独使用和组合使用)的优势。仅使用 MiSeq 读取进行组装的基因组高度准确,但缺乏连续性,通过将 MinION 读取添加到这些组装中可以部分克服这一缺陷。通过使用 MinION 读取进行初始组装,可以生成更连续的基因组组装,但这些组装更容易出错,需要进一步的打磨。当 Illumina 文库存在偏差时,情况尤其如此,就像我们的高 GC 含量和低 GC 含量的菌株一样。基因组连续性的提高极大地改善了插入序列和次级代谢生物合成基因簇的注释,这可能是因为长读长可以消除这些高度重复但对生物重要的基因组区域的歧义。
短读序列的基因组组装受到重复序列和极端 GC 含量的挑战。我们的结果表明,这些困难可以通过使用单分子、长读测序技术(如牛津纳米孔 MinION)来克服。使用 MinION 读取进行组装,然后使用 Illumina 读取进行打磨,可以生成最连续的基因组,具有足够的准确性,从而能够准确注释插入序列和次级代谢生物合成基因簇等重要但难以测序的基因组特征。因此,牛津纳米孔和 Illumina 测序的组合可以有效地推进微生物进化和基于基因组的药物发现研究。