Animal Science Department, São Paulo State University (Unesp), Jaboticabal, SP, 144884-900, Brazil.
Division of Animal Sciences, University of Missouri, Columbia, MO, 65211, USA.
BMC Genet. 2019 Jan 14;20(1):8. doi: 10.1186/s12863-019-0713-4.
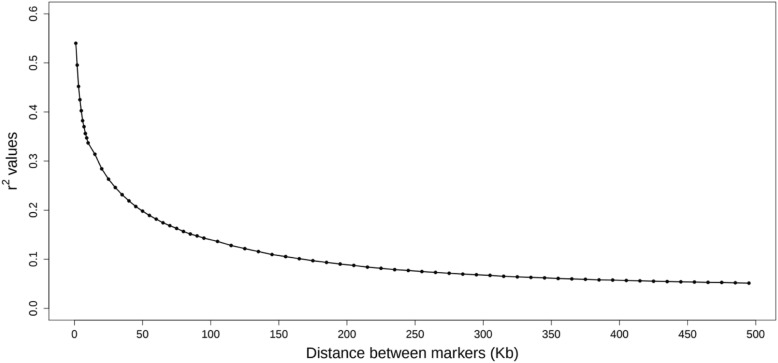
Traditional single nucleotide polymorphism (SNP) genome-wide association analysis (GWAA) can be inefficient because single SNPs provide limited genetic information about genomic regions. On the other hand, using haplotypes in the statistical analysis may increase the extent of linkage disequilibrium (LD) between haplotypes and causal variants and may also potentially capture epistastic interactions between variants within a haplotyped locus, providing an increase in the power and robustness of the association studies. We performed GWAA (413,355 SNP markers) using haplotypes based on variable-sized sliding windows and compared the results to a single-SNP GWAA using Warner-Bratzler shear force measured in the longissimus thorasis muscle of 3161 Nelore bulls to ascertain the optimal window size for identifying the genomic regions that influence meat tenderness.
The GWAA using single SNPs identified eight variants influencing meat tenderness on BTA 3, 4, 9, 10 and 11. However, thirty-three putative meat tenderness QTL were detected on BTA 1, 3, 4, 5, 8, 9, 10, 11, 15, 17, 18, 24, 25, 26 and 29 using variable-sized sliding haplotype windows. Analyses using sliding window haplotypes of 3, 5, 7, 9 and 11 SNPs identified 57, 61, 42, 39, and 21% of all thirty-three putative QTL regions, respectively; however, the analyses using the 3 and 5 SNP haplotypes, cumulatively detected 88% of the putative QTL. The genes associated with variation in meat tenderness participate in myogenesis, neurogenesis, lipid and fatty acid metabolism and skeletal muscle structure or composition processes.
GWAA using haplotypes based on variable-sized sliding windows allowed the detection of more QTL than traditional single-SNP GWAA. Analyses using smaller haplotypes (3 and 5 SNPs) detected a higher proportion of the putative QTL.
传统的单核苷酸多态性(SNP)全基因组关联分析(GWAA)可能效率低下,因为单个 SNP 提供的基因组区域遗传信息有限。另一方面,在统计分析中使用单倍型可以增加单倍型与因果变异之间的连锁不平衡(LD)程度,并且还可以潜在地捕获单倍型基因座内变异之间的上位性相互作用,从而提高关联研究的功效和稳健性。我们使用基于可变大小滑动窗口的单倍型进行 GWAA(413,355 SNP 标记),并将结果与使用 Warner-Bratzler 剪切力在 3161 头 Nelore 公牛的背最长肌中进行的单 SNP GWAA 进行比较,以确定确定影响肉质嫩度的基因组区域的最佳窗口大小。
使用单 SNP 的 GWAA 鉴定了影响 BTA3、4、9、10 和 11 上肉质嫩度的 8 个变体。然而,使用可变大小滑动单倍型窗口在 BTA1、3、4、5、8、9、10、11、15、17、18、24、25、26 和 29 上检测到 33 个假定的肉质嫩度 QTL。使用 3、5、7、9 和 11 SNP 的滑动单倍型分析分别鉴定出 57、61、42、39 和 21%的所有 33 个假定 QTL 区域;然而,使用 3 和 5 SNP 单倍型的分析,累积检测到 88%的假定 QTL。与肉质嫩度变化相关的基因参与肌发生、神经发生、脂质和脂肪酸代谢以及骨骼肌结构或组成过程。
使用基于可变大小滑动窗口的单倍型进行 GWAA 可以检测到比传统的单 SNP GWAA 更多的 QTL。使用较小的单倍型(3 和 5 SNP)进行分析可以检测到更高比例的假定 QTL。