Patel Ravi, Coulter Lucia Lee, Rimmer Joanna, Parkes Miles, Chinnery Patrick Francis, Swift Oscar
School of Clinical Medicine, University of Cambridge, Cambridge, CB2 0SP, UK.
Department of Gastroenterology, Addenbrooke's Hospital, Cambridge University Hospitals NHS Foundation Trust, Cambridge, CB2 0QQ, UK.
BMC Gastroenterol. 2019 Jan 15;19(1):11. doi: 10.1186/s12876-018-0925-5.
Mitochondrial neurogastrointestinal encephalopathy (MNGIE), due to mutations in TYMP, often presents with gastrointestinal symptoms. Two sisters, initially managed for Crohn's disease based upon clinical, imaging and pathological findings, were later found to have MNGIE. The cases provide novel clinicopathological insight, for two further reasons: both sisters remain ambulant and in employment in their late 20s and 30s; diagnosis in one sister was made after a suspected azathioprine-precipitated acute illness.
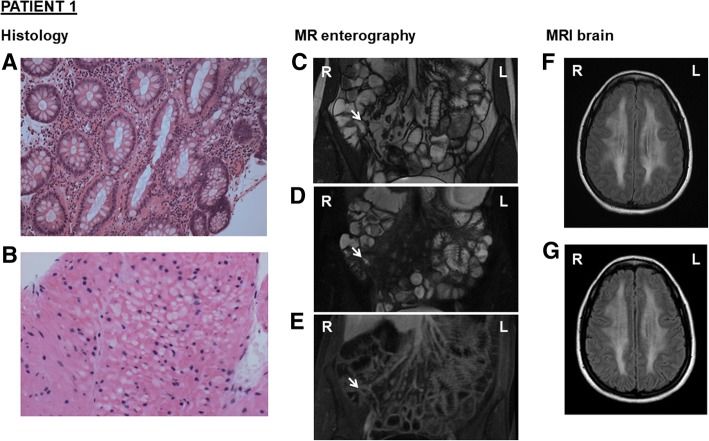
A 25-year-old female presented with diarrhoea, vomiting, abdominal pain, and bloating. Faecal calprotectin, colonic biopsies and magnetic resonance enterography were consistent with a diagnosis of Crohn's disease. Azathioprine initiation preceded admission with a sore throat, headache, myalgia, and pyrexia. Withdrawal led to rapid clinical improvement. MRI brain revealed persistent, extensive white matter changes. Elevated plasma and urine thymidine and deoxyuridine, and genetic testing for TYMP variants, confirmed MNGIE. Testing of the patient's sister, also diagnosed with Crohn's disease, revealed identical variants. In this context, retrospective review of colonic biopsies identified histological findings suggestive of MNGIE.
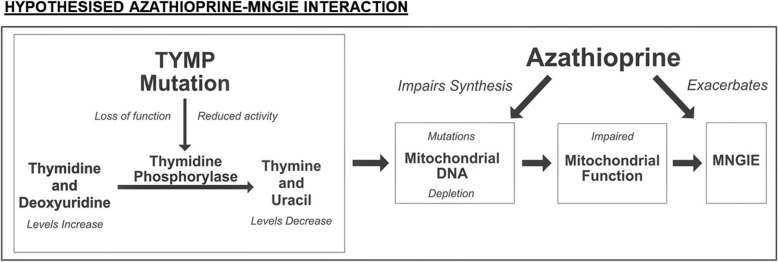
Azathioprine interference in nucleic acid metabolism may interact with the mitochondrial DNA depletion of MNGIE. Nucleotide supplementation, proposed for treatment by manipulating mitochondrial nucleoside pools, may require caution. The late onset and mild phenotype observed confirms presentation can occur later in life, and may reflect residual thymidine phosphorylase activity. Clinicians should consider measuring plasma thymidine levels in suspected Crohn's disease to rule out MNGIE, particularly if white matter abnormalities are identified on neuroimaging.
线粒体神经胃肠性脑病(MNGIE)由TYMP基因突变引起,常表现为胃肠道症状。两名姐妹最初根据临床、影像学和病理检查结果被诊断为克罗恩病,后来被发现患有MNGIE。这两个病例提供了新的临床病理见解,还有另外两个原因:两名姐妹在20多岁和30多岁时仍能行走并工作;其中一名姐妹在怀疑是硫唑嘌呤诱发的急性疾病后被确诊。
一名25岁女性出现腹泻、呕吐、腹痛和腹胀。粪便钙卫蛋白、结肠活检和磁共振肠造影结果均符合克罗恩病的诊断。入院前开始使用硫唑嘌呤,随后出现咽痛、头痛、肌痛和发热。停药后临床症状迅速改善。脑部MRI显示持续的广泛白质改变。血浆和尿液中胸苷和脱氧尿苷升高,以及对TYMP变异体的基因检测,确诊为MNGIE。对同样被诊断为克罗恩病的患者姐妹进行检测,发现了相同的变异体。在此背景下,对结肠活检进行回顾性检查发现了提示MNGIE的组织学表现。
硫唑嘌呤对核酸代谢的干扰可能与MNGIE的线粒体DNA耗竭相互作用。通过操纵线粒体核苷池进行治疗的核苷酸补充可能需要谨慎。观察到的迟发性和轻度表型证实该病可在生命后期出现,可能反映了残留的胸苷磷酸化酶活性。临床医生在疑似克罗恩病患者中应考虑检测血浆胸苷水平以排除MNGIE,特别是在神经影像学检查发现白质异常时。