Sofer Tamar, Zheng Xiuwen, Gogarten Stephanie M, Laurie Cecelia A, Grinde Kelsey, Shaffer John R, Shungin Dmitry, O'Connell Jeffrey R, Durazo-Arvizo Ramon A, Raffield Laura, Lange Leslie, Musani Solomon, Vasan Ramachandran S, Cupples L Adrienne, Reiner Alexander P, Laurie Cathy C, Rice Kenneth M
Department of Medicine, Harvard Medical School, Boston, Massachusetts.
Division of Sleep and Circadian Disorders, Brigham and Women's Hospital, Boston, Massachusetts.
Genet Epidemiol. 2019 Apr;43(3):263-275. doi: 10.1002/gepi.22188. Epub 2019 Jan 17.
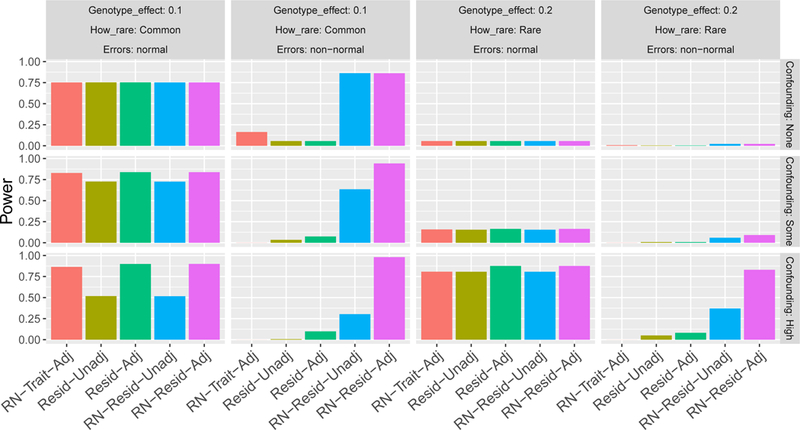
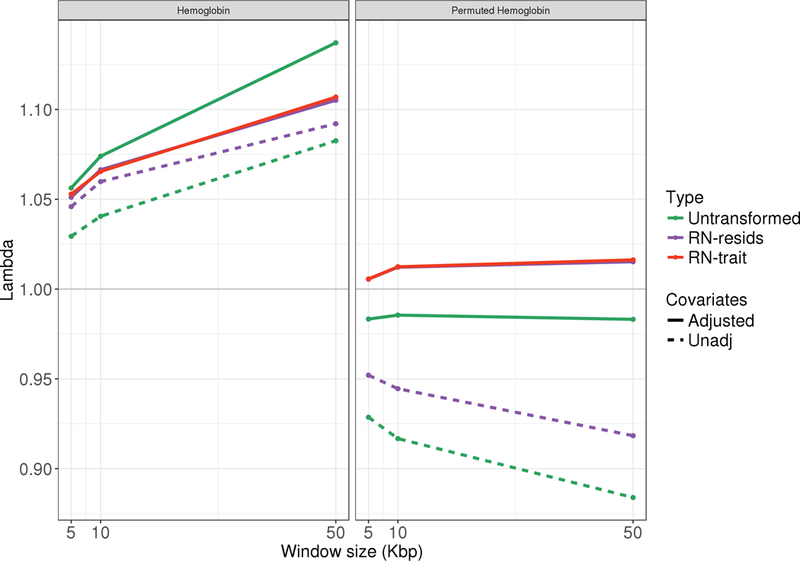
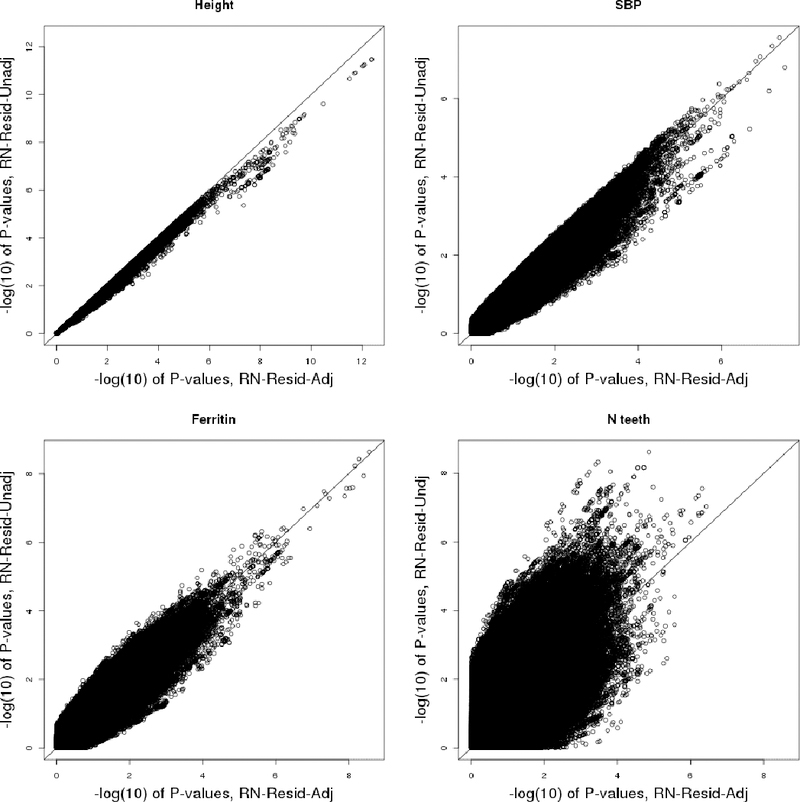
When testing genotype-phenotype associations using linear regression, departure of the trait distribution from normality can impact both Type I error rate control and statistical power, with worse consequences for rarer variants. Because genotypes are expected to have small effects (if any) investigators now routinely use a two-stage method, in which they first regress the trait on covariates, obtain residuals, rank-normalize them, and then use the rank-normalized residuals in association analysis with the genotypes. Potential confounding signals are assumed to be removed at the first stage, so in practice, no further adjustment is done in the second stage. Here, we show that this widely used approach can lead to tests with undesirable statistical properties, due to both combination of a mis-specified mean-variance relationship and remaining covariate associations between the rank-normalized residuals and genotypes. We demonstrate these properties theoretically, and also in applications to genome-wide and whole-genome sequencing association studies. We further propose and evaluate an alternative fully adjusted two-stage approach that adjusts for covariates both when residuals are obtained and in the subsequent association test. This method can reduce excess Type I errors and improve statistical power.
在使用线性回归测试基因型与表型的关联时,性状分布偏离正态性会影响I型错误率控制和统计功效,对罕见变异的影响更为严重。由于基因型预计具有较小的效应(如果有),研究人员现在通常使用两阶段方法,即首先将性状对协变量进行回归,获得残差,对其进行秩归一化,然后在与基因型的关联分析中使用秩归一化后的残差。潜在的混杂信号被认为在第一阶段已被去除,因此在实际操作中,第二阶段不再进行进一步调整。在这里,我们表明,由于错误指定的均值 - 方差关系以及秩归一化残差与基因型之间剩余的协变量关联的组合,这种广泛使用的方法可能导致具有不良统计特性的检验。我们从理论上证明了这些特性,并在全基因组和全基因组测序关联研究的应用中也进行了证明。我们进一步提出并评估了一种替代的完全调整两阶段方法,该方法在获得残差时以及在随后的关联检验中都对协变量进行调整。这种方法可以减少过多的I型错误并提高统计功效。